
Abstract
Coronaviruses cause respiratory tract infections in humans and outbreaks of deadly pneumonia worldwide. Infections are initiated by the transmembrane spike (S) glycoprotein, which binds to host receptors and fuses the viral and cellular membranes. To understand the molecular basis of coronavirus attachment to oligosaccharide receptors, we determined cryo-EM structures of coronavirus OC43 S glycoprotein trimer in isolation and in complex with a 9-O-acetylated sialic acid. We show that the ligand binds with fast kinetics to a surface-exposed groove and that interactions at the identified site are essential for S-mediated viral entry into host cells, but free monosaccharide does not trigger fusogenic conformational changes. The receptor-interacting site is conserved in all coronavirus S glycoproteins that engage 9-O-acetyl-sialogycans, with an architecture similar to those of the ligand-binding pockets of coronavirus hemagglutinin esterases and influenza virus C/D hemagglutinin-esterase fusion glycoproteins. Our results demonstrate these viruses evolved similar strategies to engage sialoglycans at the surface of target cells.
Similar content being viewed by others

Main
Coronaviruses are large, positive-sense enveloped RNA viruses in the Nidovirales order and are divided into four genera: α, β, γ and δ. Two β-coronaviruses have caused outbreaks of deadly pneumonia in humans since the beginning of the 21st century. The severe acute respiratory syndrome coronavirus (SARS-CoV) emerged in 2002 and was responsible for an epidemic that spread to five continents with a fatality rate of 10% before being contained in 2003 (with additional cases reported in 2004). The Middle East respiratory syndrome coronavirus (MERS-CoV) emerged in the Arabian Peninsula in 2012 and has caused recurrent outbreaks in humans with a fatality rate of 35%. SARS-CoV and MERS-CoV are zoonotic viruses that crossed the species barrier using bats/palm civets1 and dromedary camels2, respectively. Four other coronaviruses of zoonotic origin are endemic in the human population, accounting for up to 30% of mild respiratory tract infections and causing severe complications or fatalities in young children, the elderly and immunocompromised individuals3,4. These viruses are HCoV-NL63 and HCoV-229E (α-coronaviruses) and HCoV-OC43 and HCoV-HKU1 (β-coronaviruses). Currently, no specific antiviral treatments or vaccines are available to combat any human coronavirus. Furthermore, future cross-species transmission events of coronaviruses seem likely, given the large reservoir found in bats5,6,7. Studying coronaviruses will therefore help in understanding the principles governing cross-species transmission and adaptation to humans and in preparing for putative future zoonotic outbreaks.
Coronaviruses use homotrimers of the spike (S) glycoprotein to promote host attachment and fusion of the viral and cellular membranes for entry. S is the main antigen present at the viral surface and is the target of neutralizing antibodies during infection. As a result, it is a focus of vaccine design. S is a class I viral fusion protein synthesized as a single polypeptide chain precursor of approximately 1,300 amino acids8. For many coronaviruses, S is processed by host proteases to generate two subunits, designated S1 and S2, which remain non-covalently bound in the pre-fusion conformation. The N-terminal S1 subunit comprises four β-rich domains, designated A, B, C and D, with domain A or B acting as receptor-binding domains in different coronaviruses. The transmembrane C-terminal S2 subunit is the metastable spring-loaded fusion machinery9. During entry, S2 is further proteolytically cleaved at the S2′ site, immediately upstream of the fusion peptide10. This second cleavage step occurs for all coronaviruses and is believed to activate the protein for membrane fusion, which takes place via irreversible conformational changes11,12,13,14. In recent years, cryo-EM work led to the determination of coronavirus S glycoprotein ectodomain structures in the pre-fusion and post-fusion states, providing snapshots of the start and end points of the fusion reaction9,13,15,16,17,18,19,20,21,22,23,24. Cryo-EM structures of the SARS-CoV and MERS-CoV S glycoproteins in complex with human neutralizing antibodies also informed about the mechanism of fusion activation25.
HCoV-OC43 was isolated for the first time in 1967 from volunteers at the Common Cold Unit in Salisbury, United Kingdom. Molecular clock analysis of genome sequences suggested that HCoV-OC43 originated from a zoonotic transmission event of a bovine coronavirus (BCoV) and dated their most recent common ancestor between the 1890s and the 1950s26,27. HCoV-OC43, HCoV-HKU1, BCoV and porcine hemagglutinating encephalomyelitis virus (PHEV) use 9-O-acetyl-sialic acid (9-O-Ac-Sia) as a receptor, which is terminally linked to oligosaccharides decorating glycoproteins and gangliosides at the host cell surface28,29. The S glycoprotein of these viruses mediates 9-O-Ac-Sia binding, whereas the hemagglutinin-esterase (HE) protein acts as receptor-destroying enzyme, via sialate-O-acetyl-esterase activity, to facilitate release of viral progeny from infected cells and escape from attachment to non-permissive host cells or decoys30,31,32,33. These properties are shared with the hemagglutinin-fusion-esterase (HEF) glycoproteins of influenza C and D viruses28,34,35,36.
Sialic acids are ubiquitous terminal residues of glycoconjugates and occur in a wide variety as a result of modifications of the core N-acetyl neuraminic acid molecule and of differences in glycosidic linkages37,38,39. Previous biochemical work established that domain A of coronavirus S glycoproteins mediates attachment to oligosaccharide receptors, such as for HCoV-OC43 and BCoV, which interact with 9-O-Ac-Sia28,40,41, or MERS-CoV, which binds to α2,3-linked (and to a lesser extent to α2,6-linked) sialic acids, with sulfated sialyl-Lewis X being the preferred binder42. On the basis of the galectin-like fold of domain A of coronavirus S and mutational analyses, it was suggested that key saccharide-binding residues locate to the viral membrane distal side of the BCoV β-sandwich. Our recent work, however, indicated that the 9-O-Ac-Sia binding site of HCoV-OC43, HCoV-HKU1, BCoV and PHEV is conserved among these viruses and resides at a distinct location of domain A43. Although we validated the findings using mutagenesis and BCoV infectivity assays, no structural information is available on the mechanism of coronavirus interaction with saccharides aside from in silico modeling43. This knowledge gap limits our understanding of the roles of these receptors in viral infection or zoonosis and hinders the rational design of inhibitors.
To understand attachment of coronaviruses to sialic acids at the surface of host cells, we determined cryo-EM structures of a stabilized HCoV-OC43 S glycoprotein trimer in isolation and in complex with 5-N-acetyl,9-O-acetyl-neuraminic acid α-methyl glycoside (9-O-Ac-Me-Sia) at 2.9-Å and 2.8-Å resolution, respectively. We show that the ligand binds with fast association/dissociation kinetics in a groove on HCoV-OC43 S located at the surface of domain A. Site-directed mutagenesis combined with binding experiments validated our structural findings, and infectivity assays showed that the residues involved in 9-O-Ac-Sia binding are essential for HCoV-OC43 S–mediated entry into host cells. Our results further show that binding to free 9-O-Ac-Me-Sia and/or acidic pH did not induce fusogenic conformational changes of S, suggesting that multivalent interactions with sialoglycans and/or further attachment to a putative proteinaceous receptor44 are essential to promote membrane fusion. The receptor-interacting site is conserved in all coronavirus S glycoproteins known to attach to 9-O-Ac-sialoglycans and shares architectural similarity with the ligand-binding pockets of coronavirus HEs and influenza virus C/D HEF glycoproteins, thus highlighting common structural principles of recognition45,46.
Results
Structure of the apo-HCoV-OC43 S glycoprotein
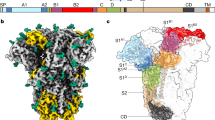
We determined a 2.9-Å resolution cryo-EM reconstruction of an apo-HCoV-OC43 S ectodomain trimer mutant, in which the S1/S2 furin cleavage site was abrogated to prevent proteolytic processing during biogenesis. HCoV-OC43 S folds as a 150-Å high and 130-Å wide compact trimer (Fig. 1a, Supplementary Fig. 1a,b and Table 1). The S1 subunit has a V-shaped architecture resulting from the 3D arrangement of its four domains (A, B, C and D), similarly to other β-coronavirus S structures9,17,18,19,20,21,22,23,24,25. The S2 subunit, which is more conserved than the S1 subunit among coronaviruses, folds as a mostly helical, elongated trimeric unit with a connector domain appended at its C-terminal end9,16 (Fig. 1a). Among available coronavirus S glycoprotein structures, HCoV-OC43 S is most similar to mouse hepatitis virus (MHV) S9 (r.m.s. deviation (r.m.s.d.) 4.7 Å over 979 aligned Cα positions) and to HCoV-HKU1 S18 (r.m.s.d. 4.5 Å over 949 aligned Cα positions), sharing 62% and 68% sequence identity, respectively. The cryo-EM reconstruction resolves 14 N-linked glycans extending from the surface of each protomer. The HCoV-OC43 S oligosaccharide density is comparable to that of SARS-CoV S and MERS-CoV S, with all three viruses belonging to the β-genus, but lower than the glycan density of the porcine delta coronavirus S (δ-genus) or the HCoV-NL63 S (α-genus) glycoproteins16,17,25.

a, Ribbon diagrams of the HCoV-OC43 S ectodomain trimer in two orthogonal orientations. The individual protomers are each in a different color, and the glycans are rendered as dark blue spheres. b, Ribbon diagrams of the superimposed HCoV-OC43 (light pink) and HCoV-HKU1 (dark gray) B domains in two orthogonal orientations. The N and C termini are labeled.
Domain B shows the highest variability within S1 subunits across coronaviruses, which correlates to the ability of different viruses to interact with distinct host receptors. For β-coronaviruses, the canonical architecture of domain B comprises a conserved five-stranded anti-parallel β-sheet, decorated with α-helices on both sides, and a highly variable external subdomain that can mediate receptor engagement for SARS-CoV19,47 or MERS-CoV48. Domain B of HCoV-OC43 and HCoV-HKU1 are structurally similar and can be superimposed with an r.m.s.d. of 1.0 Å over 251 aligned Cα positions49, with differences largely restricted to the external subdomain (Fig. 1b). The current consensus in the field is that HCoV-OC43 S does not rely on receptors other than 9-O-Ac-sialoglycans for promoting viral entry into host cells. In contrast with the MERS-CoV S and SARS-CoV S glycoproteins19,20,21,23,25,50, in which domain B adopts alternative conformations, we detected a single closed conformation of domain B in the HCoV-OC43 S structure (Fig. 1a). Only the closed domain B conformation was also observed for the MHV S9, HCoV-NL63 S16, HCoV-HKU1 S18, PDCoV S17,24 and IBV S22 glycoprotein structures.
Cryo-EM identification of a sialoside-binding site in the HCoV-OC43 S glycoprotein
HCoV-OC43, HCoV-HKU1, BCoV and PHEV attach to the surface of target cells by binding to 9-O-Ac-sialoglycans28,29. To directly visualize the binding site and characterize the molecular details of the interactions, we incubated the HCoV-OC43 S protein with 100 mM 9-O-Ac-Me-Sia, prior to vitrification and cryo-EM data collection. We determined a 3D reconstruction of the stabilized HCoV-OC43 S protein in complex with its receptor at 2.8-Å resolution, hereafter referred to as holo-HCoV-OC43 S (Supplementary Fig. 1c,d and Table 1). The resolution estimate is supported by the visible ordered water molecules interacting with the S glycoprotein, as expected at this resolution51. The structure reveals that the ligand interacts with a groove at the periphery of domain A, in agreement with the biochemical observations reported by Hulswit et al43 (Fig. 2a–c). The receptor therefore docks into a distinct groove from those used by either human galectin-3 (ref. 52) or the rhesus rotavirus sialic acid-attachment protein53 (VP8*) to recognize their respective ligands (Supplementary Fig. 2a–c).

a, Molecular surface representation of the holo-HCoV-OC43 S ectodomain trimer structure with the bound ligand shown as sticks. Protomers are individually colored. b, Surface representation of the ligand-binding site colored by electrostatic potential from −12 to +12 kBT/ec. c, Two orthogonal views of the 9-O-Ac-Me-Sia binding site. The A domain is rendered as a ribbon diagram with the side chains of surrounding residues shown as sticks. The cryo-EM density is shown as a blue mesh. In a−c, the ligand is rendered as sticks with atoms colored by element (carbon, gray; nitrogen, blue; oxygen, red). Dashed lines show a salt bridge and hydrogen bonds formed between the ligand and domain A.
The sialoside-interacting groove defines two hydrophobic pockets, designated P1 and P2 (according to the nomenclature defined by Hulswit et al43), separated by the Trp90 indole side chain, and is delineated by two loops forming the rims of the binding site, termed L1 (27-Asn-Asp-Lys-Asp-Thr-Gly-32) and L2 (80-Leu-Lys-Gly-Ser-Val-Leu-Leu-86) (Fig. 2c). The 9-O-Ac-Me-Sia C1-carboxylate forms a salt bridge with the Lys81 side chain amine and a hydrogen bond with the Ser83 side chain hydroxyl (Fig. 2c and Supplementary Fig. 3). The 5-nitrogen atom of the ligand makes a hydrogen bond with the Lys81 backbone carbonyl (Fig. 2c). The ligand N-acetyl methyl inserts into the P2 hydrophobic pocket, defined by residues Leu80, Trp90 and Phe95. The ligand 9-O-acetyl methyl docks in the P1 hydrophobic pocket, which comprises Leu85, Leu86 and Trp90, whereas the 9-O-acetyl carbonyl makes a hydrogen bond with the Asn27 side chain amide. These observations rationalize the specificity of HCoV-OC43 S for this sialoside, because the 9-O-acetyl group is accommodated by a combination of hydrogen bonding and shape complementarity (Fig. 2b,c), similarly to 9-O-Ac-Sia binding sites of coronavirus, torovirus and orthomyxovirus HEs/HEFs32,33,34,35,43,54. Although most interactions occur with the same side of the ligand, the side chain hydroxyl of residue Thr31, which faces the 9-O-Ac-Me-Sia solvent-exposed side, forms a hydrogen bond with the Trp90 indole nitrogen. This interaction participates in stapling the A domain N-terminal segment to the β-sandwich core and contributes to defining the shape of the ligand-binding groove (Fig. 2c). Overall, the ligand buries 350 Å2 of its surface upon binding to the HCoV-OC43 S protein, corresponding to approximately 62% of the 9-O-Ac-Me-Sia total solvent-accessible surface area. The observed binding mode is compatible with interactions with longer oligosaccharides, including α2,3- and α2,6-linked sialoglycans found on cell surfaces.
HCoV-OC43 S binds 9-O-Ac-Sia with fast association and dissociation rates
To characterize the binding kinetics and affinity of an individual HCoV-OC43 S binding site for 9-O-Ac-Sia receptors, we recombinantly produced the monomeric HCoV-OC43 S domain A and used biolayer interferometry to analyze its attachment to biotinylated oligosaccharides immobilized on the surface of streptavidin-coated biosensors. Domain A bound to and dissociated from 6-sialyl-5-N,9-O-acetyl-lactosamine (9-O-Ac-6SLN) with fast on and off rates. (Fig. 3a). The observed binding was specific, as it was critically dependent on the presence of the sialate-9-O-acetyl moiety, in accordance with previous observations28,32,43,55. Domain A did not detectably bind to the corresponding non-O-acetylated oligosaccharide, 6SLN. This finding is explained by the absence of the 9-O-acetyl moiety in 6SLN, which contributes one-third of the total ligand buried surface area by contacting Asn27 and the P1 pocket of the glycoprotein, as revealed in our structure (Fig. 2b,c). Moreover, binding was largely abolished by de-O-acetylation of biosensor-bound 9-O-Ac-6SLN with porcine torovirus HE (Fig. 3a). Finally, substitution of Trp90 with alanine abrogated interactions with 9-O-Ac-6SLN (Fig. 3a), thereby confirming the central role for sialoside attachment of this amino acid residue that defines the floor of the ligand-binding groove43.

a, Biolayer interferometry showing binding of wild-type or W90A monomeric HCoV-OC43 domain A to immobilized 6-sialyl-5-N-acetyl,9-O-acetyl-lactosamine (9-O-Ac-6SLN), 6-sialyl-5-N-acetyl-lactosamine (6SLN) or HE-pre-treated 6-sialyl-5-N-acetyl,9-O-acetyl-lactosamine before binding (9-O-Ac-6SLN, pre-HE) or after a successful association/dissociation event (9-O-Ac-6SLN, post-HE). b, Binding of different concentrations of wild-type monomeric A domain to immobilized 9-O-Ac-6SLN. c, Steady-state affinity determination using the curves shown in b. HCoV-OC43 A engages 9-O-Ac-6SLN with a KD = 49.7 ± 10.7 µM. d, Asn27, a key 9-O-Sia-interacting residue visualized in the holo-HCoV-OC43 S glycoprotein structure was substituted with alanine, and binding was assessed using a solid-phase lectin binding assay. Data points are averages from three independent technical triplicates. The data are normalized relative to the wild type. e, Sialoside binding to the identified site is necessary for HCoV-OC43 S-mediated entry of pseudotyped VSV-ΔG particles into host cells. n = 3 pseudovirus experiments (technical replicates). Data are normalized relative to wild type and shown as mean and s.d. of technical triplicates. f, Western-blot analysis of VSV-ΔG pseudotyped with wild-type or mutant HCoV-OC43 S. VSV-N was used as a quantitative control for the amount of virions analyzed.
Using steady-state analysis, we determined an equilibrium dissociation constant KD = 49.7 ± 10.7 µM for the HCoV-OC43 domain A−9-O-Ac-6SLN complex (Fig. 3b,c). We calculated a half-life of t1/2 = 0.7 s from the dissociation curves, a dissociation rate constant koff = 1 s−1 (koff = t1/2/ln2) and an association rate constant kon = 1.4 × 104 M−1s−1. These values predict rapid S-mediated virion attachment, particularly in high-density receptor environments such as the mucus layer, glycocalyx and cell surfaces. On the basis of these results, the mean life (1/koff) of the 1:1 complex is predicted be short, in the order of 1 s, much shorter than the mean life of an individual influenza A hemagglutinin receptor-binding domain in complex with sialic acid, which ranges between 7 and 13.5 s56. In the context of authentic virions, however, the large number of S glycoproteins at the surface of coronaviruses is likely to increase the apparent binding affinity for sialoglycans through avidity, as described for influenza virus57. We posit that HCoV-OC43 and related β-coronavirus S glycoproteins evolved to dynamically interact with host sialosides and avoid irreversible attachment to decoy receptors via HE-mediated virion elution. Dynamic binding in combination with receptor destruction could promote virion motility by directional sliding diffusion through high-density interaction sites, as recently reported for influenza A and C viruses58,59,60.
HCoV-OC43 S attachment to 9-O-Ac-sialoglycans is necessary for viral entry
Our structure rationalizes the results of our previous study in which the effect of individual HCoV-OC43 S domain A substitutions was assessed using a solid-phase lectin binding assay43. Substitution of Lys81 or Ser83 with alanine completely abrogated binding, as expected on the basis of our holo-HCoV-OC43 S structure, owing to disruption of the aforementioned electrostatic interactions with 9-O-Ac-Sia. Moreover, mutations of Leu80, Leu86 or Trp90 also disrupted binding, probably as a result of alteration of the P1 and/or P2 hydrophobic pockets accommodating the ligand 9-O-acetyl and 5-N-acetyl methyl groups, respectively. On the basis of our structure, we predicted that substitution of Asn27 with alanine would also inhibit binding, owing to loss of a hydrogen bond between the ligand 9-O-acetyl carbonyl and the Asn27 side chain amide. Using the same solid-phase lectin-interaction assays, we show that this substitution resulted in a loss of detectable binding, further validating our cryo-EM results (Fig. 3d).
We subsequently evaluated the importance of the identified interactions for HCoV-OC43 S-mediated infectivity using pseudotyped G-deficient vesicular stomatitis virus (VSV-ΔG). Substitutions at Asn27, Thr31, Leu80, Lys81, Ser83, Leu86 and Trp90 led to complete abrogation of viral entry (Fig. 3e,f), in agreement with our structural data, biolayer interferometry and solid-phase lectin binding assays, as well as the literature43. These findings (i) support the importance of the identified residues for interacting with 9-O-Ac-Sia in the context of a full-length, membrane-embedded, HCoV-OC43 S glycoprotein and (ii) indicate that attachment to oligosaccharide receptors using the binding site visualized via cryo-EM plays a critical role in promoting HCoV-OC43 S-mediated viral entry.
Free 9-O-Ac-Sia does not trigger fusogenic conformational changes
Comparison of the stabilized apo- and holo-HCoV-OC43 S glycoprotein structures did not reveal conformational rearrangements upon binding to 9-O-Ac-Sia (the two structures can be superimposed with a Cα r.m.s.d. of 0.2 Å). To validate this finding, we investigated the effect of ligand binding to wild-type HCoV-OC43 S (that is, with a native S1/S2 cleavage site sequence) in various biochemical conditions. Importantly, the HCoV-OC43 S ectodomain trimer remained uncleaved after secretion (Supplementary Fig. 4a), perhaps owing to the paucity of furin present in the secretory pathway of HEK293F cells61. Incubation of the wild-type HCoV-OC43 S ectodomain trimer with trypsin at concentrations ranging from 0.2 to 28 µg∙ml−1 (w/v), to recapitulate proteolytic priming13, led to cleavage at the S1–S2 boundary, as observed via SDS-PAGE (Supplementary Fig. 4a). Incubation with 28 µg∙ml−1 trypsin also led to cleavage of a small fraction of S at a second site, yielding a band with an apparent molecular weight of ~55 kDa, which could be consistent with cleavage at the S2′ site (Supplementary Fig. 4a), an event believed to be restricted to fusion triggering upon receptor engagement for SARS-CoV S11 or MERS-CoV S12,62. EM analysis of negatively stained samples, however, showed that the HCoV-OC43 S trimers remained in the pre-fusion conformation and were highly stable, even at the highest trypsin concentration tested (Supplementary Fig. 4b). Furthermore, we did not detect conformational changes (i) of pre-cleaved wild-type HCoV-OC43 S incubated with 100 mM 9-O-Ac-Me-Sia, (ii) after trypsin cleavage of 9-O-Ac-Me-Sia-bound wild-type HCoV-OC43 S or (iii) of pre-cleaved wild-type HCoV-OC43 S incubated at pH 4.5 (Supplementary Fig. 4c–f). Therefore, 9-O-Ac-Me-Sia binding and pH acidification of the medium, such as the one occurring in the endosomal compartment, did not trigger HCoV-OC43 S fusogenic conformational changes.
To evaluate the ability of our purified glycoprotein construct to undergo fusogenic conformational changes, we incubated the pre-cleaved wild-type HCoV-OC43 S ectodomain at 50 °C for 25 min in absence or presence of isopropanol (used to dissolve the trypsin inhibitor added to stop the proteolytic reaction) (Supplementary Fig. 4g,h). In the latter conditions, we noticed the formation of HCoV-OC43 S rosettes arising from the nonspecific interactions of multiple post-fusion trimers via the hydrophobic fusion peptides (Supplementary Fig. 4h). These biochemical conditions lowered the energy barrier between the metastable pre-fusion state and the post-fusion (ground) state, acting as a surrogate for receptor-mediated fusion activation. This finding indicated that the wild-type HCoV-OC43 S ectodomain trimer could refold to the post-fusion conformation, although neither free 9-O-Ac-Me-Sia nor pH acidification triggered this transition. It has been previously established that caveolin-mediated endocytosis is a major route of HCoV-OC43 entry into host cells63. Because we demonstrated interactions of sialoglycans with the identified site are necessary for S-mediated viral entry, we hypothesize that membrane fusion occurs upon formation of multivalent interactions with sialoglycans (via mechanical destabilization of the pre-fusion trimers) and/or binding to a putative proteinaceous receptor44, before or after virus internalization. In conclusion, 9-O-Ac-Sia-containing receptors appear to differ from the proteinaceous SARS-CoV receptor, because addition of monomeric angiotensin-converting enzyme 2 ectodomain to wild-type SARS-CoV S trimers, in the presence of trypsin, promoted refolding to the post-fusion state23,25.
A conserved sialoside attachment strategy
HCoV-OC43, BCoV, PHEV and HCoV-HKU1 are the four coronaviruses known to engage 9-O-Ac-Sia-capped sialoglycans to initiate infection of target cells. The A domain of their S glycoproteins share strikingly similar structures that can be superimposed with a Cα r.m.s.d. between 0.8 and 2.0 Å (Supplementary Fig. 5a–d).
Virtually all residues participating in interactions with 9-O-Ac-Me-Sia or the formation of the binding groove are conserved in BCoV S and PHEV S, such as Asn27, Leu80, Lys81, Leu85, Leu86, Trp90, Phe95 and Thr31 (Fig. 4a–c). Ser83HCoV-OC43, however, is substituted with Thr83BCoV/PHEV, and both side chains are expected to form a hydrogen bond with the C1-carboxylate of the ligand (Fig. 4a–c). These findings and the abrogation of BCoV and PHEV domain A–mediated hemagglutination of rat erythrocytes upon substituting Lys81/Thr83 or Trp90 with alanine43 indicate that these two viruses interact with 9-O-Ac-Sia in an identical manner to HCoV-OC43 S. The binding pocket seems to be compatible with recognition of 9-O-Ac-Sia and of 9-O-acetyl-glycolyl-neuraminic acid. Although the latter saccharide is not found in humans, it is present at the termini of oligosaccharides, decorating other mammalian and avian glycoproteins and glycolipids, and could be a receptor for BCoV and PHEV.

a–d, Zoomed-in view of the binding sites rendered as ribbon diagrams with surrounding residues shown as sticks for HCoV-OC43 (a), BCoV (b), PHEV (c), HCoV-HKU1 (d). Residues are colored by conservation, based on the analysis of all the S glycoprotein sequences available for each virus. In a, the 9-O-Ac-Me-Sia ligand is rendered as sticks with atoms colored by elements (carbon, gray; nitrogen, blue; oxygen, red). HCoV-OC43, 192 sequences; BCoV, 150 sequences; PHEV, 12 sequences; HCoV-HKU1, 28 sequences.
Many of the ligand-interacting residues or residues indirectly involved in formation of the recognition site identified in the holo-HCoV-OC43 S structure are also strictly conserved in HCoV-HKU1 S, such as Asn26HCoV-HKU1 (Asn27HCoV-OC43), Leu79HCoV-HKU1 (Leu80HCoV-OC43), Lys80HCoV-HKU1 (Lys81HCoV-OC43), Leu85HCoV-HKU1 (Leu86HCoV-OC43), Trp89HCoV-HKU1 (Trp90HCoV-OC43) and Phe94HCoV-HKU1 (Phe95HCoV-HKU1) (Fig. 4a,d), suggesting that HCoV-HKU1 S interacts with 9-O-Ac-sialoglycans using the same binding site as that identified for HCoV-OC43 S. This hypothesis is supported by site-directed mutagenesis experiments showing that substitution of Lys80HCoV-HKU1, Thr82HCoV-HKU1 (Ser83HCoV-OC43) or Trp89HCoV-HKU1 with alanine abrogated HCoV-HKU1 domain A–mediated hemagglutination of rat erythrocytes43.
Our results show that all coronaviruses recognizing host cell 9-O-Ac-sialoglycans share a conserved binding pocket and bind to the ligand via virtually identical interactions. Strikingly, BCoV HE and influenza HEF similarly interact with 9-O-Ac-Sia, despite ample differences in the architecture of their ligand-binding pockets33,34,43. Specifically, the two methyl groups of the ligand are docked into two hydrophobic depressions separated by an aromatic amino acid side chain, and hydrogen bonds are formed with the 5-nitrogen of the neuraminic acid core and the 9-O-acetyl carbonyl (Fig. 5a–c). The similarity across the three binding sites is reinforced by the observation that 9-O-Ac-Sia buries a comparable surface area at the interface with each of these glycoproteins and that the 9-O-acetyl moiety makes a major contribution to it in all three cases (~110 Å2). One notable difference, however, is that the C1 carboxylate anchors the ligand to HCoV-OC43 S via a salt bridge and a hydrogen bond, whereas it relies on the formation of one or two hydrogen bonds with the BCoV HE or influenza HEF lectin domains, respectively. These results expand on our previous biochemical work43 to demonstrate that BCoV HE and influenza HEF use structural principles similar to those of other 9-O-Ac-sialoglycan-recognizing human and animal coronaviruses for engagement to host cell receptors.

a, HCoV-OC43 S bound to 9-O-Ac-Me-Sia. b, BCoV HE bound to 5-N-acetyl-4,9-di-O-acetyl-neuraminic acid α-methylglycoside (PDB 3CL5). c, Influenza virus C HEF in complex with 9-N-Ac-Sia. In all panels, the glycoprotein is rendered as a gray surface with the bound ligand shown as sticks. The hydrogen bond formed with the carbonyl of the 9-O/N-acetyl group is shown by dashed lines.
Discussion
We structurally identified and characterized with unprecedented detail the HCoV-OC43 S sialoglycan-binding site, which is located in a groove at the surface of domain A. This site is conserved in all other coronaviruses known to attach to 9-O-Ac-Sia, including HCoV-HKU1 S (another endemic human coronavirus), and BCoV S (the presumptive zoonotic ancestor of HCoV-OC43). Our results provide a molecular framework explaining the specific recognition of 9-O-Ac-Sia-decorated oligosaccharides present at the surface of host cells targeted by these viruses. The β-sandwich architecture of domain A is conserved among all coronaviruses, and some of them feature a duplication of this domain at the S glycoprotein N-terminal region16. Other coronaviruses like MERS-CoV (β-coronavirus), infectious bronchitis virus (IBV, γ-coronavirus), porcine epidemic diarrhea virus (α-coronavirus) and transmissible gastroenteritis virus (α-coronavirus) have been described to also bind to sialoglycans (distinct from 9-O-Ac-sialosides) via their A domains during host cell infection42,64,65,66. The ligand-binding pocket identified in the holo-HCoV-OC43 S structure is not conserved in the MERS-CoV or in the IBV A domains, for which structures are available, suggesting that host attachment of this subset of viruses involve different interactions. The conserved topology of domain A among coronavirus S glycoproteins indicate that it derived from divergent evolution of an ancestral galectin domain. Viral evolution and adaptation thus lead to the use of distinct binding residues on the same domain putatively to acquire different ligand specificities such as 9-O-Ac-sialosides versus non-O-acetylated-sialoglycans. This evolutionary plasticity is reminiscent of what has been described for the BCoV HE lectin domain in comparison with influenza A/B hemagglutinin and influenza C/D HEF33,35.
Sialic acids cap numerous oligosaccharides found at the surface of eukaryotic cells and constitute an important class of receptors for several human pathogens35,37,38. Modulation of attachment to sialoglycans can therefore have profound effects on zoonotic transmission, tropism and virulence of many viruses. For instance, a single point mutation in the highly pathogenic H5N1 avian influenza virus hemagglutinin was proposed to account for most of the preference switch from avian enteric tract receptors (α2,3-linked sialic acid) to human respiratory tract receptors (α2,6-linked sialic acid)57. Although influenza A/B hemagglutinin, influenza C/D HEF and coronavirus HE have distinct architectures compared with those of coronavirus S glycoproteins, common rules of ligand engagement emerge. These rules also appear to extend to the interactions of sialoglycans with adenoviruses67 and reoviruses68. In all cases, sialic acid binding involves burying a small surface area (300–400 Å2) through contacts with a solvent-exposed groove of the protein. One face of the sialic acid ligand makes extensive interactions with the viral proteins, whereas the opposite, solvent-exposed face, makes few contacts. The binding affinity for sialic acids usually ranges between the micromolar and millimolar range, and the aforementioned viruses display numerous oligomeric spikes to enhance adsorption to target receptors through avidity57.
Despite these similarities, marked differences in the 3D organization of the binding sites explain the selectivity of different viruses for unmodified or modified sialic acids. The ligand-binding sites of BCoV HE, influenza HEF and a subset of coronavirus S glycoproteins have evolved to specifically recognize 9-O-Ac-Sia via hydrogen bonding with the 9-O-acetyl carbonyl moiety and formation of a hydrophobic pocket accommodating the 9-O-acetyl methyl32,33,34,43,54. In contrast, influenza hemagglutinin cannot accommodate 9-O-acetylated neuraminic acids, owing to steric restrictions, but a subset of hemagglutinins can bind to N-glycolyl neuraminic acids57,69. The HCoV-OC43 S, HCoV-HKU1 S, BCoV S and PHEV S glycoproteins therefore share the ligand specificity of influenza C/D HEF but are functionally more similar to influenza A/B hemagglutinin, by carrying receptor attachment and membrane fusion functions, whereas a dedicated HE (coronaviruses) or neuraminidase (influenza A/B) is responsible for the receptor-destroying activity. In conclusion, our results illuminate how coronaviruses recognize 9-O-Ac-sialosides to enable infection of susceptible cells and show that a conserved strategy is utilized to engage such ligands across coronaviruses and orthomyxoviruses.
Methods
Construct design
The fragment encoding the HCoV-OC43 S ectodomain (residues 15–1263, UniProtKB: Q696P8) was amplified by (RT-)PCR from the viral genome and placed into a modified pCAGGS mammalian expression vector with a CD5 N-terminal signal peptide (MPMGSLQPLATLYLLGMLVASVLA) and an engineered C-terminal extension encoding a GCN4 trimerization motif (IKRMKQIEDKIEEIESKQKKIENEIARIKKIK), a thrombin cleavage site (underlined) (LVPRGSLE), and an eight-residue Strep-tag (WSHPQFEK) followed by a stop codon, as previously described9,15,16,17. This construct results in fusing the GCN4 trimerization motif in register with the HR2 helix at the C-terminal end of the HCoV-OC43 S-encoding ectodomain sequence. A mutant gene carrying three R-to-G amino acid mutations to abolish the furin cleavage (754-RRSRG-758 → 754-GGSGG-758) at the S1–S2 junction (S2 cleavage site) was also generated following the same strategy. A pCAGGS vector encoding the HCoV-OC43 S domain A (residues 1–306) C-terminally extended with a thrombin cleavage site followed by the Fc region of human IgG was generated as described previously70.
Protein expression and purification
HEK293F cells were grown in suspension using FreeStyle 293 Expression Medium (Life Technologies) at 37 °C in a humidified 8% CO2 incubator rotating at 130 r.p.m. Wild-type or mutant HCoV-OC43 S ectodomain construct were transfected into 250 ml cultures with cells grown to a density of 1 million cells per milliliter using 293fectin (ThermoFisher Scientific). After 4 d, supernatant was collected, and cells were kept in culture for an additional 4 d, yielding two harvests per transfection. Recombinant wild-type or mutant HCoV-OC43 S ectodomain was purified from clarified supernatants using a 1 ml StrepTrap column (GE Healthcare). Purified proteins were concentrated and flash frozen in Tris-saline buffer (20 mM Tris, pH 8.0, 150 mM NaCl) prior to negative staining and cryo-EM analysis.
Negative stain electron microscopy
Protein samples were adsorbed to glow-discharged carbon-coated copper grids for 30 s prior to 2% uranyl formate staining. Micrographs were recorded using the Leginon software71 on a 120 kV FEI Tecnai G2 Spirit with a Gatan Ultrascan 4000 CCD camera at 67,000 nominal magnification. The defocus ranged from 1.0 to 2.0 µm, and the pixel size was 1.6 Å.
Conformational change analysis using negative-staining electron microscopy and SDS-PAGE
Wild-type HCoV-OC43 S ectodomain trimer at 1 mg∙ml−1 (6.6 μM spike monomer) was digested or not with trypsin at 14 µg∙ml−1 for 30 min at room temperature, after which 1.5 mM PMSF was added to stop the reaction. The samples were subsequently incubated: either overnight at 4 °C with 100 mM 9-O-Ac-sia; 25 min at 50 °C; or 30 min at pH 4.5 using 20 mM sodium citrate buffer before being analyzed via negative-staining EM and SDS-PAGE.
Cryo-EM sample preparation and data collection
Three microliters of HCoV-OC43 S at 1 mg∙ml−1 was applied to a 2/2 C-flat grid (Protochips) that had been glow discharged for 30 s at 20 mA. After preferential orientation was observed, 2.7 µl of HCoV-OC43 S at 10 mg∙ml−1 was mixed with 0.3 µl of n-Octyl-β-D-glucopyranoside (OG) 180 mM immediately before being applied to a glow-discharged grid. Thereafter, grids were plunge frozen in liquid ethane using an FEI Mark IV Vitrobot with a 6.5–7.5 s blot time at 100% humidity and 20 °C. Incubation of 1.1 µM HCoV-OC43 S with 100 mM 9-O-acetylated sialic acid (9-O-Ac-sia) was performed overnight at 4 °C, and immediately before vitrification, OG was added to the mixture reaction at a final concentration of 18 mM. Data were acquired using an FEI Titan Krios transmission electron microscope operated at 300 kV and equipped with a Gatan K2 Summit direct detector and Gatan Quantum GIF energy filter, operated in zero-loss mode with a slit width of 20 eV. Automated data collection was carried out using Leginon71 at a nominal magnification of 130,000× with a pixel size of 0.525 Å for apo-HCoV-OC43 S (super-resolution mode) and 1.05 Å for holo-HCoV-OC43 S (counted mode). The dose rate was adjusted to 8 counts/pixel/s, and each movie was dose-fractionated in 50 (apo) or 60 (holo) frames of 200 ms. A total of 2,211 and 2,402 micrographs were respectively collected for apo- and holo-HCoV-OC43 S, with a defocus range between 1.3 and 1.8 μm.
Cryo-EM data processing
Movie frame alignment, estimation of the microscope contrast-transfer function parameters, particle picking and extraction were carried out using Warp72. Particle images were extracted with a box size of 800 binned to 400 for apo-HCoV-OC43 S or with a box size of 400 for holo-HCoV-OC43 S, both yielding a pixel size of 1.05 Å. Reference-free 2D classification in Relion was used to parse particles from the original 197,791 and 332,912 for apo- and holo-HCoV-OC43 S, respectively. The MHV S cryo-EM map9 was used to generate an initial model of apo-HCoV-OC43 S. The initial model of holo-HCoV-OC43 S was generated using the apo-HCoV-OC43 S map. Relion 3D classification without symmetry was used to select ~83,000 and ~178,000 particles from apo- and holo-HCoV-OC43 S, respectively. CTF refinement in Relion3.0 (ref. 73) was used to refine per-particle defocus values. Particle images were subjected to the Bayesian polishing procedure implemented in Relion3.0 (refs. 73,74) before performing another round of per-particle defocus refinement. The particles were then subjected to 3D classification without refining angles and shifts using the same soft mask as that used during 3D refinement and with a tau value of 30. Final 3D refinement of the apo- and holo-HCoV-OC43 S datasets imposing C3 symmetry was carried out using non-uniform refinement in cryoSPARC75 and yielded reconstructions at 2.9- and 2.8-Å resolution, respectively. Local resolution estimation, filtering and sharpening was carried out using CryoSPARC. Reported resolutions are based on the gold-standard Fourier shell correlation (FSC) of 0.143 criterion76, and FSC curves were corrected for the effects of soft masking by high-resolution noise substitution77.
Cryo-EM model building and analysis
UCSF Chimera78 and Coot79 were used to fit the MHV atomic model (PDB 3JCL) into the holo-HCoV-OC43 S cryo-EM map. The models were subsequently manually rebuilt using Coot79. N-linked glycans were hand built into the density where visible, and the models were rebuilt and refined using Rosetta80,81,82,83. Models were analyzed using MolProbity84, Privateer85 and PISA86. Figures were generated using UCSF Chimera78 and ChimeraX87. Analysis of the ligand-binding site electrostatic surface potential was performed using PDB 2PQR88 and APBS89.
Biolayer interferometry
HCoV-OC43 S1A-Fc was expressed in HEK293T cells and purified from the cell culture supernatant by protein A chromatography, as described43. Monomeric domain A, wild type or with a W90A substitution, was subsequently obtained by on-the-bead thrombin cleavage33, after which the proteins were concentrated to up to 3.8 mg∙ml−1 in PBS, aliquoted and stored at −80 °C until further use. Biolayer interferometry analysis was performed on an Octet RED384 machine. All assays were performed using Fortebio Kinetics Buffer (KB; PBS supplemented with 0.1% BSA, 0.02% Tween20 and 0.05% sodium azide) at 30 °C. Synthetic biotinylated 6-sialyl-5-N-,9-O-acetyl-lactosamine (9OAc6SLN) or 6-sialyl-5-N-acetyl-lactosamine (6SLN) dissolved to 100 nM were loaded onto streptavidin (SA) biosensors to maximum loading levels (until no further increase in reflection was observed). Sensors were washed in KB until a stable baseline was obtained. Binding of monomeric HCoV-OC43 S domain A was performed by moving receptor-loaded sensors to wells containing 100 μl of purified protein, dissolved in KB to various concentrations, for up to 3 min, then dissociating for 3 min dissociation. To abolish unspecific binding, sensors were subjected to five successive association/dissociation cycles. To test whether binding of domain A was sialate-9-O-acetyl-dependent, biosensors loaded with 9OAc6SLN were de-O-acetylated by dipping them in wells containing 20 μg∙ml−1 porcine torovirus P4 HE-Fc45 in KB for 30 min, then washing prior to association/dissociation (pre-HE) or after a cycle of association/dissociation, upon which the biosensors were subjected to a final cycle (post-HE). The equilibrium dissociation constant, KD, was determined from three independent experiments with the ‘Response’ option of the Octet Data Analysis software. The half-life of the domain A–9OAc6SLN complex was calculated manually from the dissociation curves.
Pseudovirus entry assays
HCoV-OC43 S–pseudotyped VSV-ΔG particles were prepared as previously described43. Briefly, HEK293T cells at 70% confluency were transfected with PEI-complexed plasmid DNA. For coexpression of HCoV-OC43 S and BCoV HE-Fc, S expression vectors and pCD5-BCoV HE-Fc were mixed at molar ratios of 8:1. At 48 h after transfection, cells were transduced with VSV-G–pseudotyped VSVΔG/Fluc90 at a multiplicity of infection of 1. Cell-free supernatants were harvested at 24 h after transduction and filtered through 0.45-μm membranes, and virus particles were purified and concentrated via sucrose cushion ultracentrifugation at approximately 100,000g for 3 h. Pelleted virions were resuspended in PBS and stored at −80 °C until further use. Inoculation of HRT18 monolayers in 96-well format was performed with equal amounts of S-pseudotyped virions, as calculated from VSV-N content (roughly corresponding to the yield from 2 × 105 transfected and transduced cells), diluted in 10% FBS-supplemented DMEM. At 18 h post infection, cells were lysed using passive lysis buffer (Promega). Firefly luciferase expression was measured using a firefly luciferase assay system. Infection experiments were performed independently in triplicate, each time with three technical replicates. Pseudovirus incorporation of flag-tagged OC43 S was determined for the parental type and each of the mutants via Western blotting and by calculating the S content (measured with monoclonal antibody ANTI-FLAG M2; Sigma) relative to that of VSV-N (measured with anti-VSV-N monoclonal antibody 10G4; Kerafast).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Haagmans, B. L. et al. Middle east respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 14, 140–145 (2014).
Su, S. et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24, 490–502 (2016).
Isaacs, D., Flowers, D., Clarke, J. R., Valman, H. B. & MacNaughton, M. R. Epidemiology of coronavirus respiratory infections. Arch. Dis. Child 58, 500–503 (1983).
Menachery, V. D. et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 21, 1508–1513 (2015).
Menachery, V. D. et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl Acad. Sci. USA 113, 3048–3053 (2016).
Hu, B. et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 13, e1006698 (2017).
Bosch, B. J., van der Zee, R., de Haan, C. A. & Rottier, P. J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811 (2003).
Walls, A. C. et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114–117 (2016).
Millet, J. K. & Whittaker, G. R. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 202, 120–134 (2015).
Belouzard, S., Chu, V. C. & Whittaker, G. R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl Acad. Sci. USA 106, 5871–5876 (2009).
Millet, J. K. & Whittaker, G. R. Host cell entry of middle east respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl Acad. Sci. USA 111, 15214–15219 (2014).
Walls, A. C. et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl Acad. Sci. 114, 11157–11162 (2017).
Burkard, C. et al. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 10, e1004502 (2014).
Walls, A. et al. Crucial steps in the structure determination of a coronavirus spike glycoprotein using cryo-electron microscopy. Protein Sci. 26, 113–121 (2017).
Walls, A. C. et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat. Struct. Mol. Biol. 23, 899–905 (2016).
Xiong, X. et al. Glycan shield and fusion activation of a deltacoronavirus spike glycoprotein fine-tuned for enteric infections. J. Virol. 92, e01628-17 (2018).
Kirchdoerfer, R. N. et al. Pre-fusion structure of a human coronavirus spike protein. Nature 531, 118–121 (2016).
Kirchdoerfer, R. N. et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 8, 15701 (2018).
Pallesen, J. et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl Acad. Sci. USA 114, E7348–E7357 (2017).
Gui, M. et al. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 27, 119–129 (2017).
Shang, J. et al. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 14, e1007009 (2018).
Song, W., Gui, M., Wang, X. & Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 14, e1007236 (2018).
Shang, J.et al. Cryo-EM structure of porcine delta coronavirus spike protein in the pre-fusion state. J. Virol. https://doi.org/10.1128/JVI.01556-17 (2017).
Walls, A. C. et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion.Cell 176, 1026–1039.e15 (2019).
Vijgen, L. et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79, 1595–1604 (2005).
Lau, S. K. et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 85, 11325–11337 (2011).
Vlasak, R., Luytjes, W., Spaan, W. & Palese, P. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl Acad. Sci. USA 85, 4526–4529 (1988).
Huang, X. et al. Human coronavirus HKU1 spike protein uses O-acetylated sialic acid as an attachment receptor determinant and employs hemagglutinin-esterase protein as a receptor-destroying enzyme. J. Virol. 89, 7202–7213 (2015).
de Groot, R. J. Structure, function and evolution of the hemagglutinin-esterase proteins of corona- and toroviruses. Glycoconj. J. 23, 59–72 (2006).
Desforges, M., Desjardins, J., Zhang, C. & Talbot, P. J. The acetyl-esterase activity of the hemagglutinin-esterase protein of human coronavirus OC43 strongly enhances the production of infectious virus. J. Virol. 87, 3097–3107 (2013).
Bakkers, M. J. et al. Betacoronavirus adaptation to humans involved progressive loss of hemagglutinin-esterase lectin activity. Cell Host Microbe 21, 356–366 (2017).
Zeng, Q., Langereis, M. A., van Vliet, A. L., Huizinga, E. G. & de Groot, R. J. Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc. Natl Acad. Sci. USA 105, 9065–9069 (2008).
Rosenthal, P. B. et al. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396, 92–96 (1998).
Song, H. et al. An open receptor-binding cavity of hemagglutinin-esterase-fusion glycoprotein from newly-identified influenza D virus: basis for its broad cell tropism. PLoS Pathog. 12, e1005411 (2016).
Herrler, G. et al. The receptor-destroying enzyme of influenza C virus is neuraminate-O-acetylesterase. EMBO J. 4, 1503–1506 (1985).
Stencel-Baerenwald, J. E., Reiss, K., Reiter, D. M., Stehle, T. & Dermody, T. S. The sweet spot: defining virus-sialic acid interactions. Nat. Rev. Microbiol 12, 739–749 (2014).
Neu, U., Bauer, J. & Stehle, T. Viruses and sialic acids: rules of engagement. Curr. Opin. Struct. Biol. 21, 610–618 (2011).
Schauer, R. & Kamerling, J. P. Exploration of the sialic acid world. Adv. Carbohydr. Chem. Biochem 75, 1–213 (2018).
Peng, G. et al. Crystal structure of bovine coronavirus spike protein lectin domain. J. Biol. Chem. 287, 41931–41938 (2012).
Peng, G. et al. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl Acad. Sci. USA 108, 10696–10701 (2011).
Li, W. et al. Identification of sialic acid-binding function for the middle east respiratory syndrome coronavirus spike glycoprotein. Proc. Natl Acad. Sci. USA 114, E8508–E8517 (2017).
Hulswit, R. J. G. et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl Acad. Sci. USA 116, 2681–2690 (2019).
Collins, A. R. HLA class I antigen serves as a receptor for human coronavirus OC43. Immunol. Invest 22, 95–103 (1993).
Langereis, M. A. et al. Complexity and diversity of the mammalian sialome revealed by nidovirus virolectins. Cell Rep. 11, 1966–1978 (2015).
Bakkers, M. J. et al. Coronavirus receptor switch explained from the stereochemistry of protein-carbohydrate interactions and a single mutation. Proc. Natl Acad. Sci. USA 113, E3111–E3119 (2016).
Li, F., Li, W., Farzan, M. & Harrison, S. C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309, 1864–1868 (2005).
Lu, G. et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500, 227–231 (2013).
Ou, X. et al. Crystal structure of the receptor binding domain of the spike glycoprotein of human betacoronavirus HKU1. Nat. Commun. 8, 15216 (2017).
Yuan, Y. et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 8, 15092 (2017).
Campbell, M. G., Veesler, D., Cheng, A., Potter, C. S. & Carragher, B. A resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. eLife 4, e06380 (2015).
Seetharaman, J. et al. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J. Biol. Chem. 273, 13047–13052 (1998).
Dormitzer, P. R., Sun, Z. Y., Wagner, G. & Harrison, S. C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 21, 885–897 (2002).
Langereis, M. A., Zeng, Q., Heesters, B. A., Huizinga, E. G. & de Groot, R. J. The murine coronavirus hemagglutinin-esterase receptor-binding site: a major shift in ligand specificity through modest changes in architecture. PLoS Pathog. 8, e1002492 (2012).
Kunkel, F. & Herrler, G. Structural and functional analysis of the surface protein of human coronavirus OC43. Virology 195, 195–202 (1993).
Fei, Y. et al. Characterization of receptor binding profiles of influenza a viruses using an ellipsometry-based label-free glycan microarray assay platform. Biomolecules 5, 1480–1498 (2015).
Xiong, X. et al. Receptor binding by a ferret-transmissible H5 avian influenza virus. Nature 497, 392–396 (2013).
Guo, H. et al. Kinetic analysis of the influenza A virus HA/NA balance reveals contribution of NA to virus-receptor binding and NA-dependent rolling on receptor-containing surfaces. PLoS Pathog. 14, e1007233 (2018).
Sakai, T., Takagi, H., Muraki, Y. & Saito, M. Unique directional motility of influenza C Virus controlled by its filamentous morphology and short-range motions. J. Virol. https://doi.org/10.1128/JVI.01522-17 (2018).
Sakai, T., Nishimura, S. I., Naito, T. & Saito, M. Influenza A virus hemagglutinin and neuraminidase act as novel motile machinery. Sci. Rep. 7, 45043 (2017).
Mukherjee, S. et al. Mechanism and significance of cell type-dependent neutralization of flaviviruses. J. Virol. 88, 7210–7220 (2014).
Park, J. E. et al. Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl Acad. Sci. USA 113, 12262–12267 (2016).
Owczarek, K. et al. Early events during human coronavirus OC43 entry to the cell. Sci. Rep. 8, 7124 (2018).
Wickramasinghe, I. N., de Vries, R. P., Grone, A., de Haan, C. A. & Verheije, M. H. Binding of avian coronavirus spike proteins to host factors reflects virus tropism and pathogenicity. J. Virol. 85, 8903–8912 (2011).
Liu, C. et al. Receptor usage and cell entry of porcine epidemic diarrhea coronavirus. J. Virol. 89, 6121–6125 (2015).
Schultze, B. et al. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J. Virol. 70, 5634–5637 (1996).
Nilsson, E. C. et al. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med 17, 105–109 (2011).
Reiter, D. M. et al. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog. 7, e1002166 (2011).
Ito, T. et al. Receptor specificity of influenza A viruses correlates with the agglutination of erythrocytes from different animal species. Virology 227, 493–499 (1997).
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Tegunov, D. C., P. Real-time cryo-EM data pre-processing with Warp. Preprint at: https://www.biorxiv.org/content/10.1101/338558v1 (2018).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018).
Zivanov, J., Nakane, T. & Scheres, S.H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013).
Goddard, T. D., Huang, C. C. & Ferrin, T. E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 5, e17219 (2016).
Frenz, B. D. R. et al. Automatically fixing errors in glycoprotein structures with Rosetta. Structure 27, 1–6 (2019).
DiMaio, F., Leaver-Fay, A., Bradley, P., Baker, D. & Andre, I. Modeling symmetric macromolecular structures in Rosetta3. PLoS One 6, e20450 (2011).
DiMaio, F. et al. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. Biol. Crystallogr 66, 12–21 (2010).
Agirre, J. et al. Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 (2015).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32, W665–W667 (2004).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA 98, 10037–10041 (2001).
Kaname, Y. et al. Acquisition of complement resistance through incorporation of CD55/decay-accelerating factor into viral particles bearing baculovirus GP64. J. Virol. 84, 3210–3219 (2010).
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences (R01GM120553 to D.V.), a Pew Biomedical Scholars Award (D.V.) and an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund (D.V.), the Zoonoses Anticipation and Preparedness Initiative (IMI115760, F.A.R. and B.-J.B.), the Pasteur Institute (M.A.T. and F.A.R.), the Centre National de la Recherche Scientifique (F.A.R.), the LabEx Integrative Biology of Emerging Infectious Diseases (F.A.R.), the Netherlands Organization for Scientific Research (NWO TOP-PUNT 718.015.003, G.-J.B.) and CSC grant 2014-03250042 (Y.L.). This work was also partly supported by the Arnold and Mabel Beckman cryo-EM center and the Proteomics Resource (UWPR95794) at the University of Washington, and the Electron Imaging Center for NanoMachines supported by the NIH (1U24GM116792, 1S10RR23057 and 1S10OD018111), NSF (DBI-1338135) and CNSI at UCLA. We thank Y. Matsuura (Osaka University, Japan) for provision of the VSV-G pseudotyped VSVΔG/Fluc plasmids.
Author information
Authors and Affiliations
Contributions
M.A.T., A.C.W., Y.L., R.J.d.G. and D.V. designed the experiments. C.W. and B.-J.B. designed and cloned the protein constructs. M.A.T. and C.W. carried out protein expression and purification. Z.L. and G.-J.B. provided key reagents; M.A.T. and A.C.W. performed cryo-EM sample preparation and data collection. M.A.T., A.C.W. and D.V. processed the cryo-EM data. M.A.T. and D.V. built and refined the atomic models. Y.L. and D.K. carried out the binding and pseudovirus assays. M.A.T., A.C.W., Y.L., R.J.d.G., F.A.R. and D.V. analyzed the data. M.A.T., A.C.W. and D.V. prepared the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 CryoEM analysis of apo and holo HCoV-OC43 S glycoprotein.
a, c, Representative electron micrographs (defocus: 1.4 μm and 1.2 µm) and class averages of apo (a) and holo (c) HCoV-OC43 S trimer embedded in vitreous ice. Scale bars: 100 nm (micrograph) and 200 nm Å (class averages). b, d Gold-standard (blue) and model/map (red) Fourier shell correlation curves. The resolution was determined to 2.9 Å and 2.8 Å for the apo and holo HCoV-OC43 S glycoprotein maps, respectively. The 0.143 and 0.5 cut-off values are indicated by horizontal dashed lines.
Supplementary Figure 2 Carbohydrate ligands are recognized by distinct regions of the HCoV-OC43 S, human galectin-3 or rotavirus VP8* β-sandwich.
(a-c) Ribbon diagrams in two orthogonal orientations showing 9-O-Ac-Me-Sia bound to the HCoV-OC43 S domain A (a), galactose bound to human galectin-3 (b, PDB 1A3K) and sialic acid bound to rotavirus VP8* (c, PDB 1KQR). Each domain is shown in the same two orientations for comparison.
Supplementary Figure 3 Zoomed-in view of the sialoglycan-binding site in the holo-HCoV-OC43 S glycoprotein structure.
The A domain is rendered as a ribbon diagram with the side chains of key ligand-interacting residues shown as sticks and the corresponding cryoEM density shown as a blue mesh. The ligand is rendered as sticks with atoms colored by elements (carbon: grey, nitrogen: blue, oxygen: red).
Supplementary Figure 4 Free 9-O-Ac-Me-Sia receptor does not trigger HCoV-OC43 S fusogenic conformational changes.
(a) SDS-PAGE. (b) Purified wild-type HCoV-OC43 S ectodomain trimer in the prefusion conformation. (c-f) The wild-type HCoV-OC43 S ectodomain trimer remained in the prefusion conformation after cleavage with 28 µg.mL−1 trypsin (c), incubation with 100 mM 9-O-Ac-Me-Sia of pre-cleaved trimer (d) or trypsin cleavage of 9-O-Ac-Me-Sia-bound trimer (e), cleavage with 28 µg.mL−1 trypsin and incubation at pH 4.5 for 1 hour at room temperature (f), as visualized by single-particle electron microscopy of negatively stained samples. The wild-type HCoV-OC43 S ectodomain trimer was cleaved with 28 µg.mL−1 trypsin and heated for 20 minutes at 50 °C in the absence (g) or presence (h) of 10% isopropanol (used to dissolve the trypsin inhibitor added to stop the proteolytic reaction). Only the latter condition led to the formation of postfusion rosettes. Scale bars: 200 nm.
Supplementary Figure 5 Coronavirus S galectin-like A domains have a conserved architecture.
(a–d) Ribbon diagrams of the HCoV-OC43 (a), BCoV (b, PDB 4H14), PHEV (c, PDB 6QFY) and HCoV-HKU1 (d, PDB 5I08) A domains.
Supplementary information
Supplementary Information
Supplementary Figures 1–5
Rights and permissions
About this article

Cite this article
Tortorici, M.A., Walls, A.C., Lang, Y. et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol 26, 481–489 (2019). https://doi.org/10.1038/s41594-019-0233-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-019-0233-y
This article is cited by
-
Mannose-specific plant and microbial lectins as antiviral agents: A review
Glycoconjugate Journal (2024)
-
The role of receptors in the cross-species spread of coronaviruses infecting humans and pigs
Archives of Virology (2024)
-
Sialoglycan binding triggers spike opening in a human coronavirus
Nature (2023)
-
Development of a quantum dot-based lateral flow immunoassay strip for rapid and sensitive detection of SARS-CoV-2 neutralizing antibodies
Scientific Reports (2023)
-
Metabolic alterations upon SARS-CoV-2 infection and potential therapeutic targets against coronavirus infection
Signal Transduction and Targeted Therapy (2023)
