Article Text

Abstract
Objectives To study the responsiveness of a combined power Doppler and greyscale ultrasound (PDUS) score for assessing synovitis in biologic-naïve patients with rheumatoid arthritis (RA) starting abatacept plus methotrexate (MTX).
Methods In this open-label, multicentre, single-arm study, patients with RA (MTX inadequate responders) received intravenous abatacept (∼10 mg/kg) plus MTX for 24 weeks. A composite PDUS synovitis score, developed by the Outcome Measures in Rheumatology–European League Against Rheumatism (OMERACT–EULAR)-Ultrasound Task Force, was used to evaluate individual joints. The maximal score of each joint was added into a Global OMERACT–EULAR Synovitis Score (GLOESS) for bilateral metacarpophalangeal joints (MCPs) 2–5 (primary objective). The value of GLOESS containing other joint sets was explored, along with clinical efficacy.
Results Eighty-nine patients completed the 24-week treatment period. The earliest PDUS sign of improvement in synovitis was at week 1 (mean change in GLOESS (MCPs 2–5): −0.7 (95% CIs −1.2 to −0.1)), with continuous improvement to week 24. Early improvement was observed in the component scores (power Doppler signal at week 1, synovial hyperplasia at week 2, joint effusion at week 4). Comparable changes were observed for 22 paired joints and minimal joint subsets. Mean Disease Activity Score 28 (C reactive protein) was significantly reduced from weeks 1 to 24, reaching clinical meaningful improvement (change ≥1.2) at week 8.
Conclusions In this first international prospective study, the composite PDUS score is responsive to abatacept. GLOESS demonstrated the rapid onset of action of abatacept, regardless of the number of joints examined. Ultrasound is an objective tool to monitor patients with RA under treatment.
Trial registration number NCT00767325.
- Rheumatoid Arthritis
- Ultrasonography
- Disease Activity
- DMARDs (biologic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Synovitis is the first manifestation of rheumatoid arthritis (RA), which ultimately leads to joint erosion.1 Recommendations for management of RA have evolved to include early aggressive therapy to prevent or delay permanent structural damage of the joints.2 ,3 This has fuelled a search for more accurate methods to monitor early changes in synovitis.
Power Doppler and greyscale ultrasound (termed PDUS in this paper) is a promising, non-invasive imaging method to assess synovitis in RA: results from numerous studies suggest that it provides additional information to clinical and conventional radiographic examinations.4–8 PDUS has demonstrated similar sensitivity and specificity to MRI in detecting synovitis,9 ,10 and may provide a less daunting option for patients and a more accessible alternative to MRI in clinical practice. PDUS is also able to evaluate multiple joints in a short period of time.
Various scoring systems have been proposed for the evaluation of synovitis by PDUS, at both individual joint and patient levels.11 Such scoring systems vary with respect to the definition of ultrasound-detected synovitis and Doppler and greyscale methods used, and there is no consensus regarding the number and specific joints evaluated. Regardless, data from published studies indicate that PDUS scoring systems could be useful in monitoring early response to biological treatment in patients with RA, independently of the correlation with other clinical outcomes.12–20
The Outcome Measures in Rheumatology-Ultrasound (OMERACT-US) Task Force, with funding received from the European League Against Rheumatism (EULAR), works to standardise the use of ultrasonography in RA and has developed a composite scoring system (the OMERACT–EULAR composite PDUS score) to detect and score synovitis. This score combines greyscale-assessed synovial hyperplasia (SH) with intrasynovial power Doppler (PD) signal for evaluating synovial activity. The score has demonstrated validity and intraobserver and interobserver reliability in cross-sectional datasets, applicability to all joints and consistency between machines.21–24
This report presents data from the first prospective international study designed to assess the capability of the composite PDUS score to measure the early effect and time course of response to treatment with abatacept in biologic-naïve patients with active RA despite methotrexate (MTX). The primary objective of this study was to investigate the responsiveness of the composite PDUS score, as defined by an improvement in the global score (Global OMERACT–EULAR Synovitis Score, or GLOESS) of the bilaterally evaluated second to fifth metacarpophalangeal joints (MCPs 2–5). Other study objectives included the responsiveness of GLOESS with the full set of 22 paired joints, in order to then identify the minimum subset of joints (ie, reduced set) that can adequately monitor disease activity at the patient level, and the clinical efficacy and safety of abatacept plus MTX in this patient population.
Methods
Study design
APPRAISE (NCT00767325) was a 24-week, phase IIIb, open-label, multicentre, single-arm study conducted at 21 sites across Europe (Denmark, France, Germany, Hungary, Italy, Norway, Spain and the UK).
All patients received intravenous abatacept at a weight-tiered dose of 10 mg/kg at baseline (day 1), and at weeks 2, 4, 8, 12, 16, 20 and 24, in addition to stable doses of concomitant MTX (≥15 mg/week). MTX dose increases were not permitted, and MTX dose decreases were allowed only in cases of intolerance. Oral corticosteroid use (stable dose of ≤10 mg prednisone/day) was permitted. Patients could take non-steroidal anti-inflammatory drugs or other rescue analgesics if required, except in the 12 h before each visit. Concomitant biological agents or other disease-modifying antirheumatic drugs were not permitted.
The study was approved by the Institutional Review Board/Independent Ethics Committee and local ethics committees, and was conducted in accordance with the ethical principles underlying the European Union Directive 2001/20/EC and the United States Code of Federal Regulations on good clinical practice, as defined by the International Conference on Harmonisation. All patients provided written informed consent.
Patients
Eligible patients were ≥18 years of age, had American College of Rheumatology (ACR), defined RA (1987 classification criteria) for at least 6 months and were receiving MTX (≥15 mg/week) for at least 3 months prior to baseline, with a stable MTX dose for at least 28 days before baseline (except in cases of intolerance). Patients were required to have active disease, defined by a baseline Disease Activity Score (DAS28 (C reactive protein, CRP)) of >3.2 or tender joint count (TJC) and swollen joint count (SJC) of ≥6 and a CRP level of greater than the upper normal limit. Patients previously treated with a biological disease-modifying antirheumatic drug were excluded.
Assessments
PDUS assessment
At screening and baseline, all patients underwent a bilateral PDUS examination of MCPs 2–5. An OMERACT–EULAR composite PDUS score of ≥2 for at least two MCPs and ≥1 for at least one other MCP joint was required for inclusion. At baseline, and weeks 1, 2, 4, 6, 8, 12, 16, 20 and 24, PDUS was performed bilaterally for 22 joint sites (MCPs 1–5, proximal interphalangeal joints (PIPs) 1–5, wrist, elbow, shoulder (glenohumeral), knee, ankle (tibiotalar), hind foot (talonavicular and calcaneocuboidal) and metatarsophalangeal joints (MTPs) 1–5). Medium-level to high-level ultrasound machines were used (Esaote Technos MPX, MyLab 70, Toshiba Aplio, GE Logic (series 5, 7 and 9) or Siemens Acuson Antares), employing high-frequency (12–18 MHz) transducers. Doppler parameters were adjusted according to the device used (range of pulse repetition frequency 400–800 Hz; Doppler frequency 7–11.1 MHz). PDUS evaluation was performed at each site by an independent expert in musculoskeletal ultrasound, blinded from clinical evaluations. There were no changes in ultrasound settings during the study, and no upgrading of software. Centres were advised to create a fixed study setting, to be used at each evaluation. All images were recorded, anonymised and sent for central reading; this allowed random verification of the constancy of settings over time.
The PDUS assessment consisted of an evaluation of hypoechoic SH using greyscale and of synovial vascularisation using PD. The prespecified set of 22 paired joints was scanned at each visit on the dorsal aspect, with the joint in a neutral position, except for the knee, which was also examined in a flexed position (30°). Standardised joint and probe positions were used, based on a reference atlas, which also showed examples of synovitis grading for each joint examined.
The presence of synovitis (ie, SH with or without PD) was scored according to the OMERACT–EULAR PDUS composite semiquantitative scale (0–3; box 1).21 GLOESS scores for MCPs 2–5 of both hands (primary efficacy assessment) and for the 22 paired joints were then calculated using the sum of the composite PDUS scores for all joints examined, giving a potential score of 0–24 for MCPs 2–5 and of 0–132 for the 22 paired joints. Each single component of joint inflammation (SH, PD) was also scored separately at each visit, using semiquantitative scales (0–3; box 1). Joint effusion (JE) was also scored separately using a semiquantitative scale (see online supplementary data).
Ultrasound scoring systems at joint and patient levels
Joint level (for individual joints)
A) Single components
Greyscale inflammatory (hypoechoic) synovial hyperplasia
Grade 0: no hypoechoic synovial hyperplasia
Grade 1: minimal hypoechoic synovial hyperplasia (filling the angle between the periarticular bones, without bulging over the line linking tops of the bones)
Grade 2: hypoechoic synovial hyperplasia bulging over the line linking tops of the periarticular bones but without extension along the bone diaphysis
Grade 3: hypoechoic synovial hyperplasia bulging over the line linking tops of the periarticular bones and with extension to at least one of the bone diaphyses
PD signal
Grade 0: no flow in the hypoechoic synovial hyperplasia
Grade 1: up to three single spots signals or up to two confluent spots or one confluent spot plus up to two single spots
Grade 2: vessel signals in less than half of the area of the synovium (≤50%)
Grade 3: vessel signals in more than half of the area of the synovium (>50%)
B) Composite score
OMERACT–EULAR composite PDUS synovitis score
Grade 0 (normal joint): no greyscale-detected synovial hyperplasia and no PD signal
Grade 1 (minimal synovitis): grade 1 synovial hyperplasia and ≤ grade 1 PD signal
Grade 2 (moderate synovitis): grade 2 synovial hyperplasia and ≤ grade 2 PD signal; or grade 1 synovial hyperplasia and a grade 2 PD signal
Grade 3 (severe synovitis): grade 3 synovial hyperplasia and ≤ grade 3 PD signal; or grade 1 or 2 synovial hyperplasia and a grade 3 PD signal
Patient level
Global OMERACT–EULAR Synovitis Score
Sum of composite PDUS scores for all joints assessed (eg, for MCPs 2–5, global PDUS score would range from 0 to 24)
MCP, metacarpophalangeal joint; OMERACT–EULAR, Outcome Measures in Rheumatology–European League Against Rheumatism; PD, power Doppler; PDUS, PD and greyscale ultrasound.
The completion of a training session and examinations was required by all PDUS assessors in order to be qualified for PDUS evaluation in this study and ensure homogeneity of synovitis scoring. Quality control assessment of the scoring for MCPs 2–5 was performed for the first patient from each centre and at the end of the study using a randomised sample of images from each site. In cases of discordance, centralised readings were used.
Clinical evaluations
Disease activity was evaluated at baseline (day 1), weeks 1, 2, 4, 6, 8, 12, 16, 20 and 24 using DAS28. Mean change in DAS28 from baseline, the proportion of patients achieving clinically meaningful improvements (CMIs) in DAS28 (defined as improvement from baseline of ≥1.2), and the proportions of patients achieving a DAS28 <2.6, DAS28 ≤3.2 or achieving ACR/EULAR Boolean remission22 (TJC28 ≤1, SJC28 ≤1, CRP ≤1 mg/dL and patient global assessment (0–10 scale) ≤1) were calculated.
Safety evaluations
Adverse events (AEs), serious AEs and events of special interest (infections, serious infections, autoimmune events, malignancies and acute infusion reactions) were assessed in all patients receiving at least one dose of study drug, and included events occurring up to 56 days after the last infusion.
Statistical analysis
This was an exploratory study, and therefore, the sample size was not based on statistical power calculation. No data were available from longitudinal multicentre studies using the OMERACT–EULAR composite PDUS score. Based on similar studies using ultrasound to measure responsiveness to biological agents in patients with RA, the sample size was set at 100 patients; the width of the 95% CI of the estimated improvement in GLOESS for MCPs 2–5 was estimated to be <0.98 on each side of the interval.
All patients with a PDUS examination at baseline and with at least one postbaseline efficacy assessment were included in the efficacy analyses. The time point with the earliest sign of improvement in synovitis (defined as the time point for which ‘0’ was not included in the 95% CIs for the mean change from baseline in GLOESS (MCPs 2–5) at that and all later time points) was recorded. Last observation carried forward was applied for patients who discontinued prior to week 24.
Descriptive statistics were provided by time point for changes from baseline in GLOESS and for all efficacy and safety variables. For continuous data, time point estimates, SE and two-sided 95% CIs were calculated. For categorical data, frequencies and percentages were provided. Proportions and two-sided 95% CIs were calculated for dichotomous categorical data.
Principal component analysis (PCA) was used to identify a reduced set of joints that best represented the GLOESS for the 22 paired joints. The PCA was performed separately at baseline, week 12 and week 24 to identify two good subsets at each time point, explaining at least 75% of the total variation of the GLOESS over the three time points.23 The best subset was identified using the efficiency measure criteria. Effect size, expressed as standardised response means (SRMs) of GLOESS and components, was investigated for each of the joint sets based on mean changes from baseline to week 24.
Results
Patient disposition and baseline characteristics
One hundred and four patients were enrolled in this study. The majority of patients were female, with long disease duration and high disease activity, in addition to ultrasound evidence of synovitis at baseline (table 1). Eighty-nine patients (86%) completed the 24-week open-label treatment period (figure 1).
Baseline demographics and clinical characteristics

Patient disposition for all patients who received at least one dose of study drug (N=104). Owing to compliance issues (major deviation due to the lack of participation in training), patients from one site (n=8) were excluded before power Doppler and greyscale ultrasound analyses were performed.
PDUS findings
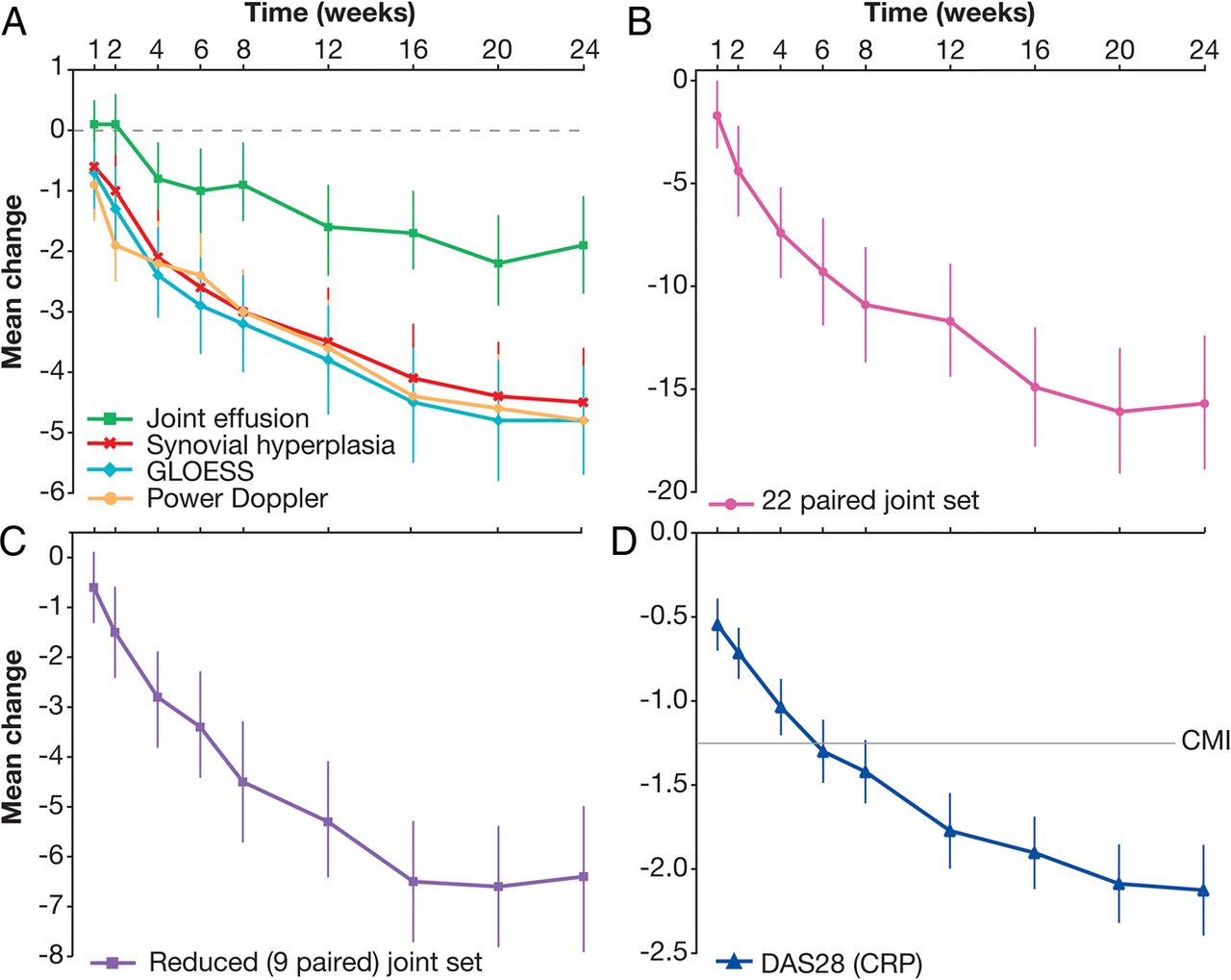
Owing to compliance issues (major deviation owing to the lack of participation in PDUS training), patients from one site (n=8) were excluded, and PDUS analyses performed using data from the remaining 96 patients. Figure 2A–C shows the responsiveness of PDUS throughout the study period. The earliest PDUS sign of improvement in synovitis was observed as early as week 1; with a mean (95% CI) change from baseline in GLOESS (MCPs 2–5) of −0.7 (−1.2 to −0.1). In the assessment of individual joint synovitis, the composite PDUS score and PD signal showed similar trends, with statistically significant reductions by week 1. SH showed significant reduction by week 2 and JE showed a first statistically significant sign of improvement at week 4. Continued improvements in GLOESS (MCPs 2–5) and in each of the component scores were observed up to week 24 (figure 2A). However, smaller improvements were observed for JE over the 24-week treatment period compared with GLOESS, PD and SH scores (figure 2A). Similar trends in the component scores were observed when using the full 22 paired joint evaluation (data not shown).
{kind=link}
{kind=link}
Mean change in (A) GLOESS and the component scores for metacarpophalangeal joints 2–5, (B) GLOESS for the 22-paired joint set, (C) GLOESS for the reduced (nine-paired) joint set and (D) DAS28 (CRP) >24 weeks of treatment with abatacept plus methotrexate. Error bars represent 95% CIs. Data are for patients with baseline and at least one postbaseline assessment, with last observation carried forward. PDUS analyses were completed for 96 of the 104 patients, as patients from one site (n=8) were excluded due to compliance issues. (A) CI did not cross zero from week 1 for GLOESS: −0.7 (−1.2 to −0.1) or power Doppler: −0.9 (−1.5 to −0.3); CI did not cross zero from week 2 for synovial hyperplasia: −1.0 (−1.6 to −0.4); CI did not cross zero from week 4 for joint effusion: −0.8 (−1.4 to −0.3). (B) CI did not cross zero from week 1 −1.7 (−3.4 to −0.1). (C) CI did not cross zero from week 2 −1.5 (−2.4 to −0.6). (D) CI did not cross zero from week 1 −0.5 (−0.7 to −0.4). CMI, clinically meaningful improvement (ie, improvement from baseline in DAS28 score of ≥1.2); CRP, C reactive protein; DAS, disease activity score; GLOESS, Global OMERACT–EULAR (Outcome Measures in Rheumatology–European League Against Rheumatism) Synovitis Score; PDUS, power Doppler and greyscale ultrasound.
Reduced joint set
In our population, the reduced joint set that best represented the composite PDUS score for 22 paired joints over three time points (baseline, week 12 and week 24), determined by PCA, comprised nine paired joints (including both large and small joints: shoulder, elbow, wrist, MCP1, MCP4, PIP2, knee, MTP3 and MTP5). Similar onset and time course of improvement in GLOESS were observed for the 22-paired, nine-paired and MCP (2–5) joint sets (figure 2A–C).
To assess the consistency of this new reduced joint set, its responsiveness was compared with reduced joint sets reported by Naredo et al24 and Backhaus et al.25 The Backhaus (unilateral) joint set was adapted to be assessed bilaterally in this study, as the dominant hand was not known during PDUS assessment. Mean changes from baseline to week 24 in the nine paired joint GLOESS and components, and the associated SRM were comparable with the other two reduced joint sets (table 2).
Comparison of reduced nine-paired joint set with existing six-paired and seven-paired joint sets: change in GLOESS and components
Clinical efficacy assessments
Clinical efficacy analyses were carried out for all 104 patients. Mean DAS28 was significantly reduced by week 1, and continued to decrease throughout the 24-week study period (figure 2D), reaching the threshold for CMI at week 8. In patients with DAS28 measurements available at both week 12 and week 24 (n=98), the proportion of patients achieving DAS28 ≤3.2 or DAS28 <2.6 increased over the 24-week treatment period: 40% (39/98) and 19% (19/98), respectively, at week 12, compared with 57% (56/98) and 41% (40/98) at week 24. Furthermore, the more stringent ACR/EULAR Boolean criteria for remission were achieved by 11% of patients at week 24, with an additional 22% of patients achieving three of the four criteria for Boolean remission at week 24.
No correlations were observed at any time point between changes from baseline in DAS28, in GLOESS or component scores for any joint set, or using just the 28 joints used for DAS28 (see online supplementary material for data). At all time points, for any joint set, correlations were also very low between GLOESS or the scoring of each single component and the SJC and TJC (either separate or combined, see online supplementary material). However, we observed the same trend in response whatever the measure used (PDUS or DAS): the higher the PDUS, GLOESS and DAS at baseline, the less frequently Low Disease Activity Score (LDAS) or low GLOESS were achieved at 6 months. This was also seen for changes over time in both measures when expressed as continuous variables.
Safety
AEs were reported in 60% of the total population, of which 21% were assessed as possibly, probably or certainly related to study drug. Safety with abatacept was consistent with previous findings, with no unexpected AEs, deaths or opportunistic infections reported. A safety summary is in the online supplementary material.
Discussion
This was the first international multicentre study to document the responsiveness of PDUS in patients with RA starting treatment with abatacept plus MTX. It also validated the composite PDUS scoring system in this setting. The responsiveness of this scoring system was able to show statistically significant improvement as early as week 1. The same responsiveness was observed using DAS28, although changes in mean DAS28 did not reach the threshold for CMI until week 8.
The composite score incorporates both PD and greyscale measures of synovitis, evaluating changes in both activity and morphology of synovitis. Both PD and SH scores contributed to responsiveness. As expected, JE showed the lowest sensitivity to change following treatment, with the earliest sign of improvement observed at week 4. This is in agreement with the idea that effusion alone does not reflect a principal part of synovial activity and is less responsive to treatment. Therefore, JE was not included in the calculation of the OMERACT–EULAR composite PDUS score.
This study has limitations. First, the single-arm design precludes an assessment of the efficiency of PDUS in comparing treatment groups with different levels of response; such an assessment can be performed only within a randomised clinical trial. Second, the open label design could lead to bias from a positive expectation of response. However, such an expectation would likely affect clinical measurements more strongly than ultrasound, leading to larger clinical improvements compared with ultrasound—a phenomenon not seen in this study. Other factors that might influence the response would influence both measurement modalities, and thus not affect the comparison between clinical and ultrasound response. Furthermore, the sample size of 100 patients, extrapolated from a previous longitudinal study using a similar ultrasound scoring system 16 and the decision to analyse GLOESS in MCPs 2–5 as the primary efficacy outcome (owing to their frequent involvement in RA), may have resulted in suboptimal power. As discussed, the sensitivity of synovitis scores based on greyscale and/or PD signal can be affected by variables relating to the technology used, and by factors such as ambient temperature, transducer pressure26 and patient positioning.27 Clinical characteristics may also be relevant; for example, a low level of inflammation at baseline might make it harder to measure response to treatment. Finally, although several joint sets were compared in this study, the influence of using different joints with more or less PD activity (eg, MCP vs talocrural joint) on GLOESS remains unclear.
This study has identified a new reduced set of nine paired joints to represent the comprehensive 22 paired joint GLOESS. Onset and time course of improvement of this new set was similar to other sets, including previously published reduced joint sets.24 ,25 Of note, we adapted a previously published 7-joint score (which evaluated only the dominant side)25 to a bilateral paired joint set, which may have increased its sensitivity. If confirmed, bilateral evaluation of a reduced joint set could replace a full-body assessment, making such evaluation more suitable for use in routine clinical practice.25
The early onset of response to treatment with abatacept (1 week) in biologic-naïve patients with RA is in agreement with findings from the ASSET (IM101-119: Impact of intravenous Abatacept on Synovitis, osteitis and Structural damage in patients with rheumatoid arthritis and an inadequate response to mEthotrexate: a randomized controlled Trial) and AMPLE (IM101-235: Abatacept versus adaliMumab comParison in bioLogic-naïvE rheumatoid arthritis (RA) subjects with background methotrexate) studies.28 ,29 The safety profile is consistent with the long-term profile demonstrated for the pooled intravenous abatacept clinical trial experience,30 and no new safety signals were identified.
In conclusion, this study documents the responsiveness of the OMERACT–EULAR composite PDUS score in biologic-naïve patients with RA starting abatacept, with changes detectable as early as week 1. A newly proposed nine-paired joint set worked as well as a 22-paired joint set for monitoring disease. An accompanying article explores these data further, by investigating if the composite PDUS score can be used to predict clinical response in this patient population, and if it can differentiate between early and late clinical responders. Further work is also required to test the PDUS scoring system and proposed joint count in other RA cohorts.
Acknowledgments
The authors would like to thank the OMERACT–EULAR-US Task Force and principal investigators of the APPRAISE study. APPRAISE principal investigators: Silvano Adami, Vivi Bakkenheim, HBH, Stefano Bombardieri, M-AD’A, Paul Emery, Liana Euller-Ziegler, Gianfranco Ferraccioli, Maurizio Galeazzi, Philippe Gaudin, Walter Grassi, AI, Herbert Kellner, Thierry Lequerré, IM, EN, MØ, Fredeswinda Romero, Istvan Szombati, Lene Terslev, Jacqueline Uson, Esther Vicente, OV and RJW. The authors also thank Wendy Kerselaers, Harry Goyvaerts and Nathalie Schmidely from Bristol-Myers Squibb, Coralie Poncet from DOCS International and Christel Perrone from the CRO ICON for their contribution to the design, analysis and conduct of the study. Professional medical writing and editorial assistance was provided by Laura McDonagh, PhD, at Caudex and was funded by Bristol-Myers Squibb.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Hans WJ Bijlsma
CG at the time of study.
Funding The APPRAISE study and subsequent statistical analyses were funded and performed by Bristol-Myers Squibb, which was involved in the design of the study; in the collection, analysis and interpretation of data; in the writing of this manuscript; and in the decision to submit the manuscript for publication.
Competing interests M-AD’A has received speaker's bureau fees from Bristol-Myers Squibb, AbbVie, UCB, MSD and Roche Pharma, as well as research grants from Pfizer. HBH received honoraria for scientific lectures from AbbVie, Roche, BMS, Pfizer and MSD, and received grants for scientific studies from AbbVie, Roche and Pfizer. IM has received consulting fees from Bio Iberica Pharma, AbbVie, GE and ESAOTE. EN has received consulting fees from AbbVie, Roche Pharma, Bristol-Myers Squibb, Pfizer, UCB, General Electric and Esaote, as well as research grants from MSD. MØ has received personal fees from AbbVie, BMS, Boehringer-Ingelheim, Celgene, Eli Lilly, Janssen, Genmab, GSK, Merck, Pfizer, Regeneron, Roche, Sanofi and UCB; grants from AbbVie, Merck and UCB; and non-financial support from AbbVie, BMS, Janssen, Merck, Pfizer, Roche and UCB. MB has received consulting fees from Bristol-Myers Squibb and personal fees from Mundipharma, Roche, Pfizer, GSK and Novartis. CG is a former employee of Bristol-Myers Squibb. KVM and MLB are employees of Bristol-Myers Squibb.
Ethics approval Hopital Ambroise Pare, Comité de Protections des Personnes Idf Vlll Lab D’Anatomopathologie, 9 Ave Charles De Gaulle, Boulogne-Billancourt 92100, France. Comitato Etico Per La Sperimentazione Del Farmaco, Asur, Territoriale 5 Di Jesi Via Gallodoro 68, Jesi (An) 60035, Italy. Azienda Ospedaliera Universitaria Policlinico G. Martino, Comitato Etico Scientifico Via Consolare Valeria, 1, Messina 98124, Italy. Leeds East Research Ethics Committee, Yorkshire and Humber Rec Cntr Off. First Floor, Millside Mill Pond Lane, Leeds, England LS6 4RA, United Kingdom. Ceic Fundacio Unio Catalana D’Hospitals, Area De Serveis C/Bruc 72-74 1a, Barcelona 08009, Spain. Azienda Universitaria Senese, Comitato Etico Locale La Sperimentazione Clinica Dei Medicinali Farmacia Aous—Viale Bracci, Siena 53100, Italy. De Videnskabsetiske Komiteer For Region Hovedstaden, Kongens Vaenge 2, Hillerod 3400, Denmark. Com Sperimentazione Clin Dei Med Azienda Osp-Univ Pisana, Via Roma 67, Pisa 56126, Italy. Comitato Etico Dell’Universita Cattolica Del Sacro Cuore, Policlinico Universitario Agostino Gemelli Di Roma Largo A Gemelli 8, Roma 00168, Italy. Rek Sorost, Frederik Holsts Hus Ulleval Terrasse Ulleval Sykehus Kirkeveien 166, Oslo 0450, Norway. Ceic—Fundacion Jimenez Diaz-Ute, Avda. Reyes Catolicos, 2-2a, Madrid 28040, Spain. Hospital Mostoles, Ceic Area 8 C/Rio Jucar S/N, Madrid 28935, Spain. Universita Di Roma La Sapienza, Comitato Etico Dell’Azienda Policlinico Umberto I, Roma 00161, Italy. Ceic Area 9-Hosp Severo Ochoa De Leganes Y Hosp De Fuenlabrad, Avenida Orellana S/N Leganes, Madrid 28911, Spain. Hospital Universitario La Princesa, C/Diego De Leon, 62, Madrid 28006, Spain. Farmakologiai Etk Bizottsaga, Arany J.U. 6-8., Budapest 1051, Hungary. Comitato Etico Per La Sperimentazione Dell’Azienda, Ospedaliera Istituti Ospitalieri Di Verona Piazzale Stefani 1, Verona 37126, Italy. Ethikkommission Der Ludwig-Maximilians Universitaet, Marchioninistr. 15, Muenchen 81377, Germany.
Provenance and peer review Not commissioned; externally peer reviewed.