Article Text

Abstract
Objectives Autophagy is a homeostatic process to recycle dispensable and damaged cell organelles. Dysregulation of autophagic pathways has recently been implicated in the pathogenesis of various diseases. Here, we investigated the role of autophagy during joint destruction in arthritis.
Methods Autophagy in osteoclasts was analysed in vitro and ex vivo by transmission electron microscopy, Western blotting and immunohistochemistry for Beclin1 and Atg7. Small molecule inhibitors, LysMCre-mediated knockout of Atg7 and lentiviral overexpression of Beclin1 were used to modulate autophagy in vitro and in vivo. Osteoclast differentiation markers were quantified by real-time PCR. The extent of bone and cartilage destruction was analysed in human tumour necrosis factor α transgenic (hTNFα tg) mice after adoptive transfer with myeloid specific Atg7-deficient bone marrow.
Results Autophagy was activated in osteoclasts of human rheumatoid arthritis (RA) showing increased expression of Beclin1 and Atg7. TNFα potently induced the expression of autophagy-related genes and activated autophagy in vitro and in vivo. Activation of autophagy by overexpression of Beclin1-induced osteoclastogenesis and enhanced the resorptive capacity of cultured osteoclasts, whereas pharmacologic or genetic inactivation of autophagy prevented osteoclast differentiation. Arthritic hTNFα tg mice transplanted with Atg7fl/fl×LysMCre+ bone marrow cells (BMC) showed reduced numbers of osteoclasts and were protected from TNFα-induced bone erosion, proteoglycan loss and chondrocyte death.
Conclusions These findings demonstrate that autophagy is activated in RA in a TNFα-dependent manner and regulates osteoclast differentiation and bone resorption. We thus provide evidence for a central role of autophagy in joint destruction in RA.
- Arthritis
- Rheumatoid Arthritis
- TNF-alpha
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that results in severe destruction of articular cartilage and bone.1 Osteoclasts are the major cellular mediators of bone degradation in RA, which results in articular erosions and systemic osteoporosis. In this context, osteoclast precursors and mature osteoclasts are abundant at sites of arthritic bone erosions.2 ,3 The differentiation of osteoclasts from its precursors, so-called osteoclastogenesis, depends on the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL).4–6 In addition, proinflammatory cytokines, such as tumour necrosis factor α (TNFα) can enhance the osteoclast differentiation.7 Indeed, mice overexpressing human TNFα (hTNFα tg) show increased osteoclastogenesis and destructive arthritis closely resembling RA.8 ,9
Autophagy describes a general mechanism, by which cells degrade unnecessary or dysfunctional cellular organelles through the lysosomal machinery.10 The degradation of cellular organelles can maintain cellular energy levels and guarantee cell survival during starvation, but may turn into cell death if nutrient supply cannot be restored.10 Proper regulation of autophagy guarantees the fine balance of synthesis, degradation and recycling of cellular components. These functions render autophagy essential for proper cell differentiation and organ development.11
Autophagy is initiated by the formation of an isolation membrane, which then elongates to enclose the target organelles and to form the so-called autophagosome. The autophagosome subsequently fuses with lysosomes to degrade the intravesicular components. Several key regulators of autophagy have been identified in the last years. Beclin1 is essential for the initiation of autophagy.12–15 Atg7 mediates the elongation of the isolation membrane, which culminates in conversion of a soluble form of microtubule-associated protein one light chain three (LC3-I) to phosphatidylethanolamine-conjugated membrane-bound form (LC3-II), a process often used to demonstrate active autophagy.16
Although autophagy is tightly regulated to limit uncontrolled activation, deregulated autophagy may contribute to the pathogenesis of various diseases including infections, cancer, neurodegenerative and heart disease.17 Of particular interest, a most recent study showed that autophagy regulates the release of lysosomal components by osteoclasts.18 This finding may have profound impact on understanding the mechanisms of bone damage in arthritis, and in finding new strategies for prevention and treatment.
In this study, we aimed to elucidate the role of autophagy in human RA and in murine inflammatory arthritis, and the function of autophagy in TNFα-mediated bone destruction. We demonstrated that autophagy is activated in RA in a TNFα-dependent manner and stimulates osteoclast differentiation. By contrast, selective inhibition of autophagy in monocytes strongly reduces osteoclast differentiation and joint destruction in hTNFα tg mice. Together, these findings provide evidence for a central role of autophagy in joint destruction in RA.
Materials and methods
Mice and bone marrow transplantation
For bone marrow transplantation, 4-week-old recipient hTNFα tg mice (strain Tg197; C57BL/6 background)8 were irradiated with 10.5 Gy (Stabilipan; Siemens, Erlangen, Germany). After 24 h, bone marrow was reconstituted by intravenous injection of either 5×106 Atg7fl/fl× LysMCre− BMCs, or Atg7fl/fl× LysMCre+ BMCs, or BMCs transduced with lentiviruses encoding Beclin1 (LVBeclin1), or lentiviral control vectors (LVscramble).19 ,20 The animal studies were approved by the local animal welfare committee.
Isolation of osteoclast precursors and osteoclast differentiation assay
Bone marrow-derived osteoclasts were prepared as previously described.21 In brief, donor mice were sacrificed at the age of 6–8 weeks. BMCs from tibia and femur were flushed with PBS and filtered through a 0.4 μm filter. The cells were cultured in α-MEM (Invitrogen, Carlsbad, California, USA) containing 10% fetal bovine serum. One day after plating, floating cells were harvested in complete medium with 20 ng/ml murine M-CSF (rmM-CSF) and 1–30 ng/ml murine RANKL (R&D Systems, Ambington, UK) for 3 days. Medium and cytokines were replaced after 72 h. Osteoclast differentiation was evaluated by staining cells for tartrate-resistant acid phosphatase (TRAP) using a Leukocyte Acid Phosphatase Kit (Sigma-Aldrich, St Louis, Missouri, USA).
Lentiviral infection
After 72 h of stimulation with M-CSF and RANKL, bone marrow-derived osteoclasts were infected with either LV-Beclin1 or LVscramble19 at a multiplicity of infection of 10 for 24 h.
In vitro bone resorption assay
BMCs were plated on bone slices (IDS, London, Great Britain) at a density of 5×104 cells/slice in 96-well culture plates with 200 µl culture medium per well. The cultured medium was exchanged every third day. After 14 days, bone pits were stained with 1% toluidine blue O (Sigma-Aldrich).
Clinical assessment
Clinical evaluation was performed weekly and was performed in a blinded manner as described.22 Briefly, paw swelling was examined in all four paws, and a clinical score of 0–3 was assigned as follows: 0=no swelling, 1=mild swelling, 2=moderate swelling and 3=severe swelling of the toes and ankle. In addition, grip strength was examined in each paw, using a 3 mm diameter wire, and was scored on a scale from 0 to −4 as follows: 0=normal grip strength, −1=mildly reduced, −2=moderately reduced, −3=severely reduced, and −4=no grip strength. Mice were weighed weekly.
Histomorphometric analysis
Decalcified, paraffin-embedded hind paws were cut in 5 μm sections and stained with toluidine blue, haematoxylin (Merck, Darmstadt, Germany) or TRAP, using a leukocyte acid phosphatase staining kit (Sigma-Aldrich). Bone erosion, osteoclast number, and cartilage destruction were quantified with a Zeiss Axioskop 2 microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a digital camera and image analysis system (OsteoMeasure; Osteometrics, Decatur, Georgia, USA).23
Immunofluorescence staining
Synovial tissue sections from patients with RA and osteoarthritis (OA) were incubated with polyclonal rabbit anti-Atg7 (AnaSpec, San Jose, California, USA) or Beclin1 (Abcam, Cambridge, UK). Irrelevant isotype antibodies were used as negative controls. Polyclonal goat anti-rabbit antibodies labelled with fluorescent dye Alexa Fluor 488 (Invitrogen, Darmstadt, Germany) were used as secondary antibodies. Quantification was performed with ImageJ software (V.1.41; National Institutes of Health) and the intensity of immunofluorescence was analysed under equal conditions for all samples within the experiments. This includes equal exposure times and light intensity. The fluorescence intensity of 5–10 randomly selected osteoclasts per sample was quantified by Image J.
Western blot analysis
Whole-cell lysates were prepared as previously described.24 Polyvinylidene fluoride membranes were incubated with polyclonal rabbit anti-Atg7 (AnaSpec), Beclin1 (Abcam) or LC3 (Novus, Jena, Germany). Horseradish peroxidase-conjugated polyclonal goat anti-rabbit or monoclonal rabbit anti-mouse antibodies (DAKO, Hamburg, Germany) were used as secondary antibodies. Equal loading of proteins was confirmed by β-actin (Sigma-Aldrich).
Quantitative real-time PCR
Gene expression was quantified by SYBR Green real-time PCR as described.24 The following primer pairs were used: mouse cathepsin K: 5′-GGAAGAAGACTCACCAGAAGC-3′ and 5′-GTCATATAGCCGCCTCCACAG-3′; mouse TRAP 5′-CGACCATTGTTAGCCACATACG-3′ and 5′-TCGTCCTGAAGATACTGCAGGTT-3′; mouse OSCAR 5′-TCGCTGATACTCCAGCTGTC-3′ and 5′-ATCCCAGGAGTCACAACTGC-3′; mouse nuclear factor of activated T cells c1 (NFATc1), 5′-CAACAAGCGCAAGTACAGTCTC-3′ and 5′-CAGGTATCTTCGGTCACACTGA-3′; mouse β-actin, 5′-TGGCATTGTGGAAGGGCTCATGAC-3′ and 5′-ATGCCAGTGAGCTTGCCGTTCAGC-3′. Samples without enzyme in the reverse transcription reaction (Non-RT-controls) were used as negative controls to exclude genomic contamination. β-actin was used to normalise for the amounts of loaded cDNA. Differences were calculated with the threshold cycle (Ct) and the comparative Ct method for relative quantification.
MicroCT analysis
Hind paws were analysed by microCT (μCT) (μCT35; SCANCO Medical AG, Brüttisellen, Switzerland). The following acquisition parameters were used: voltage: 40 kV; x-ray current: 250 μA; exposure time: 5000 ms/projection, 720 projections; matrix: 1024×1024; and voxel size in reconstructed image: 9 μm.
Transmission electron microscopy
For transmission electron microscopy, cells were harvested and fixed with 2.5% glutaraldehyde in cacodylate buffer solution (ph 7.4, containing sucrose). Thereafter, cells were washed, postfixed with 2% osmium tetroxide in cacodylate buffer and embedded in 2% agarose. During dehydration by ethanol, the cells were fixed with 1.0% uranyl acetate in 70% ethanol, followed by embedding in araldite and cutting with an ultramicrotome (Reichert Ultracut, Vienna, Austria). Images were captured with a Zeiss EM10 electron microscope (Carl Zeiss AG) and a Gatan SC1000 OriusTM CCD camera in combination with the Digital Micrograph TM software (Gatan GmbH, Munich, Germany).
Statistical analysis
All data are presented as median with IQR using the GraphPad Prism 4.0 Software. Differences between the groups were tested for their statistical significance by non-parametric Mann–Whitney U test. A p value of less than 0.05 was considered statistically significant.
Results
Atg7 and Beclin1 are overexpressed in RA osteoclasts
We first analysed whether autophagy is activated in human osteoclasts of RA patients. Indeed, the expression of Atg7, which is essential for the elongation of the isolation membrane during autophagy, was increased in TRAP-positive cells of RA patients compared with samples from OA patients (figure 1A). The expression of Beclin1, which stimulates the initiation of autophagy, was also increased in TRAP-positive cells of RA patients (figure 1B). The average fluorescence intensity was increased by 77% for Atg7 and by 284% for Beclin1 (p=0.002 and 0.004; figure 1C). Together, these results suggest that autophagy is activated in osteoclasts in RA.

Atg7 and Beclin1 are overexpressed in osteoclasts of patients with RA. Overexpression of Atg7 and Beclin1 in osteoclasts indicates activated autophagy at sites of bone erosions in RA patients. (A, B) Representative images of osteoclasts in synovial tissue from patients with RA and OA double-stained for tartrate-resistant acid phosphatase (TRAP) and either Atg7 or Beclin1 are shown at 1000-fold magnification. TRAP stainings (purple) are shown in the upper panels. Immunofluorescence staining for either Atg7 (A) or Beclin1 (B) (both green) are shown in the lower panels. Scale bar: 10 μm, n=6 each. (C, D) Relative fluorescence intensity of Atg7 (C) or Beclin1 (D) was determined by Image J. **indicates p<0.01. This figure is only reproduced in colour in the online version.
TNFα stimulates autophagy in murine osteoclasts in vitro and in vivo
Given the key role of TNFα in inflammatory bone resorption in arthritis, we hypothesised that TNFα might stimulate autophagy in RA osteoclasts. Indeed, we found that TNFα upregulated the expression of Atg7 and Beclin1 with increased mRNA and protein levels of both autophagy markers in murine osteoclasts, in particular, under conditions with lower concentrations of RANKL (1 ng/ml) that may mimic onset of inflammation or less active disease. The stimulating effect of TNFα on Atg7 and Beclin1 was less pronounced in the presence of very high concentrations of RANKL (30 ng/ml) (figure 2A–C).

TNFα induces autophagy in osteoclasts in vitro and in vivo. (A) Representative Western blots of lysates from the mature osteoclasts stimulated with receptor activator of NF-κB ligand (RANKL) (1 ng/ml), RANKL + TNFα (1 ng/ml, 40 ng/ml), RANKL + TNFα (1 ng/ml, 200 ng/ml) or RANKL (30 ng/ml) using anti-Atg7, anti-Beclin1, anti-LC3 I, II, and anti-β-actin antibodies for detection (n = 3 for each). (B, C) Atg7 and Beclin1 mRNA levels in the osteoclasts were analysed by real-time PCR. n = 3 for each. (D, E) Representative images of joint sections from wildtype or hTNFα tg mice double-stained with tartrate-resistant acid phosphatase (TRAP) and the autophagy markers Atg7 (D) or Beclin1 (E). TRAP staining for osteoclasts (purple) is shown in the upper panels of both figures. Immunofluorescence staining with Atg7 or Beclin1 (green) as indicated by arrows is shown in the lower panels. Scale bar: 15 μm, n = 6 for each group. *indicates p 0.05 and **indicates p 0.01. This figure is only reproduced in colour in the online version.

Autophagy regulates receptor activator of NF-κB ligand-induced osteoclast differentiation. Activation of autophagy by lentiviral overexpression of Beclin1 stimulates osteoclast differentiation, whereas, inhibition of autophagy, either by knockout of Atg7 or by treatment with bafilomycin A1, prevents osteoclast differentiation. (A) Representative electron microscopy images of osteoclasts derived from Atg7fl/fl×LysMCre− BMCs or Atg7fl/fl×LysMCre+ BMCs are shown at a 12 000-fold magnification. Scale bar: 0.5 μm. Note the mitochondrion engulfed in an autophagosome with typical double membrane in osteoclasts derived from Atg7fl/fl×LysMCre− and electron-dense immature autophagic vacuoles in BMCs derived from Atg7fl/fl×LysMCre+. (B–D) Representative images of tartrate-resistant acid phosphatase (TRAP) stainings from in vitro-differentiated osteoclasts derived from BMCs derived from Atg7fl/fl×LysMCre− or Atg7fl/fl×LysMCre+ mice (B), treated with either Bafilomycin A1 or its solvent DMSO (C), or infected with lentiviral Beclin1, or the control scramble lentivirus (D), respectively, are shown (n=5 for each setting) at a 100-fold magnification. The relative number of osteoclasts defined as TRAP-positive cells with more than three nuclei in each setting are shown in the graphs in the middle. The relative area covered by osteoclasts (mean from 100 osteoclasts per setting) is presented in the panels on the right. (E–G) Quantification of mRNA levels of NFATc1, OSCAR, TRAP and CatK in indicated osteoclasts derived from BMCs as well as previously described (B, C and D, respectively). n=5 For each group in each setting. *indicates p<0.05; **indicates p<0.01. This figure is only reproduced in colour in the online version.
Consistent with the induction of Atg7 and Beclin1, TNFα also stimulated the conversion of LC3 I into LC3 II in osteoclasts, which is another hallmark of active autophagy. To confirm the stimulatory effects of TNFα on autophagy in vivo, we analysed the expression of Atg7 and Beclin1 in osteoclasts of hTNFα tg mice and wildtype littermates. The expression levels of Atg7 and Beclin1 were increased by 160% and 380% in osteoclasts of hTNFα tg mice compared with the findings in TRAP-positive cells in sections of control mice (p=0.007 and 0.009; figure 2E,F). Together, these results demonstrate that TNFα activates autophagy in murine osteoclasts in vitro and in vivo.
Autophagy regulates murine osteoclast differentiation and bone resorption in vitro
To determine, whether autophagy plays a role for osteoclast differentiation, we first performed electron microscopy (figure 3A). We observed numerous autophagosomes containing mitochondria and other organelles in differentiating osteoclasts generated from BMCs of wildtype mice. In contrast, mature autophagocytic vacuoles engulfing mitochondria and other organelles were absent in Atg7-deficient osteoclasts, but electron-dense immature autophagosomes and lysosomes accumulated in the cytoplasm of Atg7-deficient cells (figure 3A). We therefore hypothesised that the impaired degradation of damaged organelles, together with the accumulation of immature autophagolysosome, might interfere with osteoclastogenesis. Indeed, inhibition of autophagy by genetic or pharmacologic approaches prevented osteoclast differentiation. Osteoclast differentiation was reduced in bone marrow cells derived from Atg7fl/fl×LysM Cre+ with decreased numbers of osteoclasts and decreased mean size of osteoclasts as compared with Atg7fl/fl×LysM Cre− cells. Similar results were obtained upon incubation of wildtype bone marrow cells with bafilomycin, a chemical inhibitor of autophagy (figure 3B,C).
To investigate whether activation of autophagy enhances the potential of monocytes to differentiate into osteoclasts, we overexpressed Beclin1 using a lentiviral construct. Overexpression of Beclin1 potently induced autophagy in murine osteoclasts (see online supplementary material figure S1A). Activation of autophagy significantly increased the number of multinucleated, TRAP-positive osteoclasts. In addition, the mean size of osteoclasts increased upon overexpression of Beclin1 (figure 3D).
We next evaluated whether modulation of autophagy alters the expression of osteoclast-associated genes. Consistent with the effects on osteoclast numbers and size, overexpression of Beclin1 increased the mRNA levels of NFATc1, OSCAR, TRAP and cathepsin K (figure 3E–G). By contrast, deficiency of Atg7, or treatment with bafilomycin, a selective inhibitor of autophagy, decreased the expression of osteoclast-associated genes.
To further elucidate the effects of autophagy on osteoclast function, we performed in vitro bone resorption assays. Activation of autophagy significantly enhanced the resorptive capacity of murine osteoclasts, whereas genetic or pharmacologic inhibition of autophagy dramatically reduced bone resorption in vitro (figure 4). To distinguish between effects mediated by decreased osteoclast counts and effects of impaired osteoclast function, we increased the number of BMCs by twofold to compensate for decreased osteoclast numbers due to impaired osteoclastogenesis. After confirming equal numbers of osteoclasts in all settings with this approach, we repeated the bone resorption assays using those modified conditions. We found that despite similar numbers of osteoclasts, bone resportion was still decreased in Atg7fl/fl×LysM Cre+ cells or cells treated with Bafilomycin A1, respectively, as compared with Atg7fl/fl×LysM Cre− and dimethyl sulfoxide-treated controls. However, the differences were less pronounced then in previous experiments, in which equal numbers of BMCs were seeded at the beginning of the experiment. Together, those findings demonstrate that decreased function and impaired differentiation both contribute to the reduced bone resorbing capacity.

Autophagy regulates osteoclast-mediated bone resorption in vitro. (A–C) Representative images of in vitro bone resorption assays from murine BMCs infected with LV-scramble (con) or LV-Beclin1 (A), BMCs derived from Atg7fl/fl × LysMCre− or Atg7fl/fl × LysMCre+ mice (B) and BMCs treated with the solvent DMSO or Bafilomycin A1 (C), respectively, are shown on the left at 200-fold magnification. The area of bone resportion for each setting is presented on the right; n = 6 *indicates p<0.05; **indicates p<0.01. This figure is only reproduced in colour in the online version.
Local bone erosion and osteoclast numbers are decreased in hTNFα tg mice transplanted with Atg7fl/fl×lysM Cre+ BMCS
To determine the effects of autophagy on TNFα-induced arthritis and bone destruction, hTNFα tg mice were transplanted with bone marrow cells from Atg7fl/fl× LysM Cre+ mice to selectively target monocytes in hTNFα tg model (Atg7fl/fl× Cre+→hTNFα tg mice). We first confirmed that the depletion of Atg7 occurred selectively in monocytes of Atg7fl/fl×LysM Cre+, but not in other cell types, such as fibroblasts (see online supplementary figure S1B). The effective inhibition of autophagy in monocytic cells of Atg7fl/fl×LysM Cre+ mice was demonstrated by Western blots for Atg7, Beclin, LC 3 I and LC3 II (see online supplementary figure S1C).
Clinical signs of inflammation, such as paw swelling, grip strength and body weight did not differ between Atg7fl/fl× LysM Cre+→hTNFα tg mice and Atg7fl/fl×LysM Cre−→hTNFα tg mice. We also did not observe differences in the inflamed area by histomorphometry (figure 5), indicating that selective inhibition of autophagy in monocytic cells does not affect joint inflammation in TNFα-induced arthritis.

Joint inflammation is not ameliorated in hTNFα tg mice transplanted with Atg7fl/fl×LysMCre+ BMCs. (A) Representative H&E staining images of the tarsal joints from Atg7fl/fl×LysM Cre− → hTNFα or Atg7fl/fl×LysM Cre+ → hTNFα tg mice are showed at a 100-fold magnification. N = 6 for each group. (B) Histomorphometric quantification of the inflamed area in the tarsal joints demonstrated no differences between Atg7fl/fl×LysM Cre− → hTNFα and Atg7fl/fl×LysM Cre+ → hTNFα tg mice. (C–E) Clinical parameters of inflammation, such as paw swelling (C), grip strength (D) and body weight (E) did not differ between both groups of mice. NS = not significant. This figure is only reproduced in colour in the online version.
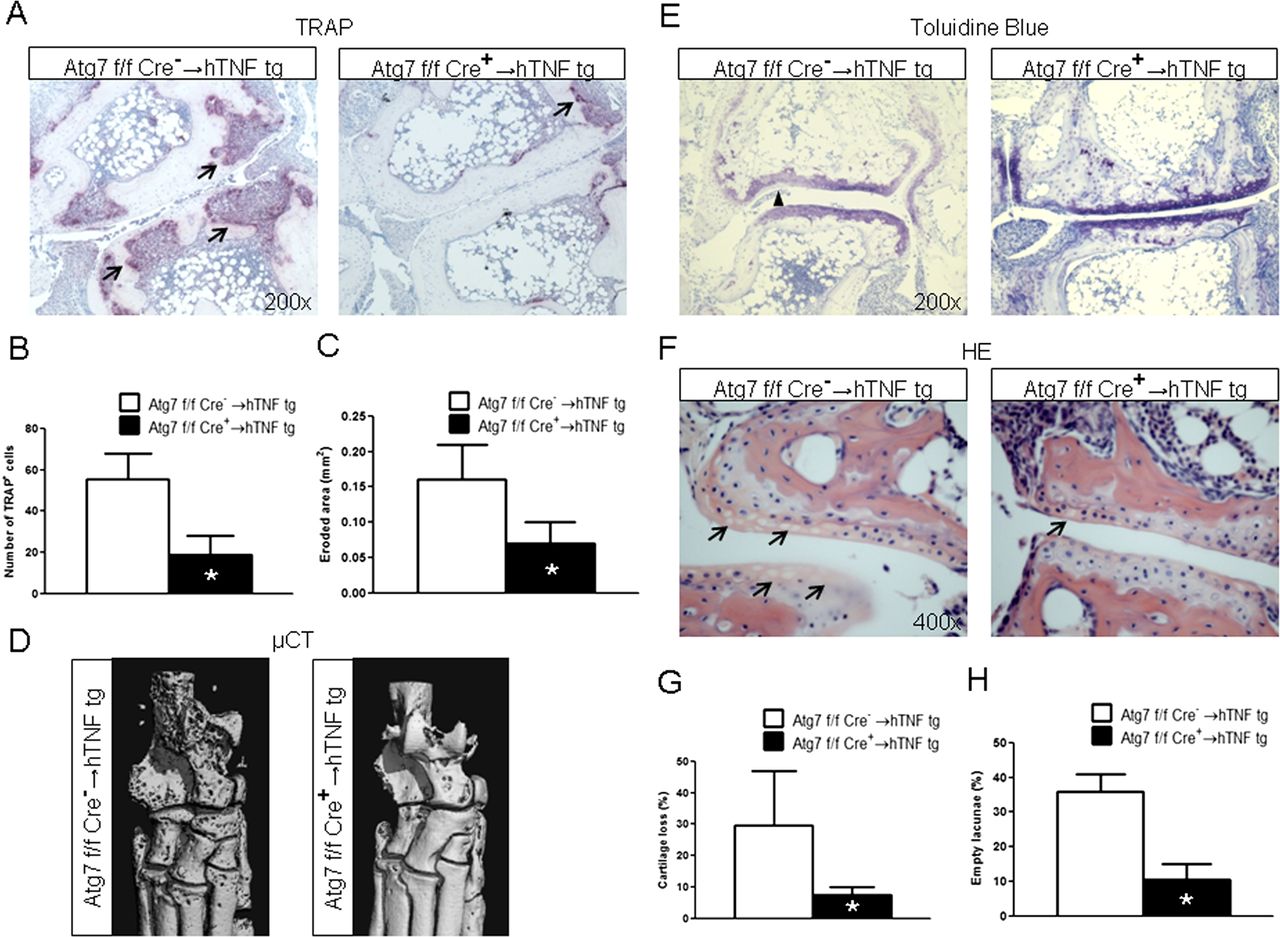
We next investigated whether targeting of autophagy affects structural damage in hTNFα tg mice. Indeed, the osteoclast-covered area and the number of osteoclasts was strongly reduced in Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with hTNFα tg mice reconstituted with Atg7fl/fl×LysM Cre− BMC (Atg7fl/fl×LysM Cre− → hTNFα tg mice) (figure 6A,B). The eroded area was also significantly reduced in Atg7fl/fl×LysM Cre+ → hTNFα tg mice (figure 6C). Consistently, µCT demonstrated that Atg7fl/fl×LysM Cre+ → hTNFα tg were largely protected from TNFα-induced bone destruction with significantly reduced bone erosions (figure 6D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Selective deletion of Atg7 in monocytic cells prevents structural damage in hTNFα tg mice. (A) Representative tartrate-resistant acid phosphatase stainings of sections from tarsal joints of hTNF mice 5 weeks after BMCS transplantation are shown at a 200-fold magnification. (B) Histomorphometric analyses of the osteoclast-covered surface demonstrated a significant reduction in Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with Atg7fl/fl×LysM Cre− → hTNFα mice. (C) The eroded area was also significantly decreased in Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with Atg7fl/fl×LysM Cre− → hTNFα mice. (D) µCT Confirmed the decreased bone erosions in Atg7fl/fl×LysM Cre+ → hTNFα tg mice. One representative image for each group of mice is shown; scale bar: 0.5 mm. (E) Representative toluidine blue stained sections demonstrate the reduced proteoglycan loss in Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with Atg7fl/fl× LysM Cre− → hTNFα mice. Increased proteoglycan loss within cartilage is indicated by arrowheads in sections stained with toluidine blue (200-fold magnification). (F) Histomorphometric quantification demonstrated significant differences in proteoglycan loss. (G) Representative H&E-stained sections with prominent empty lacunae in Atg7fl/fl×LysM Cre− → hTNFα mice, but not in Atg7fl/fl×LysM Cre+ → hTNFα mice (400-fold magnification). (H) Significantly decreased numbers of empty lacunae in Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with Atg7fl/fl×LysM Cre− → hTNFα tg mice. n = 6 For each group in all experiments. *indicates p 0.05. This figure is only reproduced in colour in the online version.
We also analysed non-transgenic mice transplanted with bone marrow from Atg7fl/fl LysM Cre+ or Atg7fl/fl LysM Cre− mice. Consistent with the role of TNFα as a potent inducer of autophagy, we did not observe differences in osteoclast numbers or bone structure in non-TNFα transgenic mice (data not shown).
Deficiency of Atg7 with subsequent inhibition of autophagy prevented also TNFα-mediated proteoglycan loss as shown by Toluidine blue staining and reduced numbers of empty lacunae (figure 6E–H). To further elucidate the mechanism by which LysM Cre driven knockdown of Atg7 may protect from cartilage loss, we analysed the expression levels of the monocyte/macrophage-derived cytokines IL-1β and IL-6, both of which have been implicated in cartilage destruction in arthritis. The levels of IL-1β and IL-6 were both significantly decreased in serum of Atg7fl/fl×LysM Cre+ → hTNFα tg mice compared with Atg7fl/fl×LysM Cre− → hTNFα tg mice (see online supplementary figure S3). Thus, the decreased expression of IL-1β and IL-6 may contribute to the observed protection of cartilage in Atg7fl/fl×LysM Cre+ → hTNFα tg mice.
Discussion
TNFα is a key player in the pathogenesis of RA that orchestrates synovial inflammation and bone degradation. In the present study, we demonstrate that TNFα stimulated autophagy in osteoclasts. In detail, TNFα induced the expression of Atg7 and Beclin1, two essential mediators of autophagy, and stimulated the conversion of LC3 I to II, another hallmark of autophagy. Consistent with these in vitro findings, Atg7 and Beclin1 were upregulated in osteoclasts of hTNFα tg mice. Autophagy was also activated in patients with RA with increased staining for Atg7 and Beclin1 in osteoclasts at sites of bone erosion. Our findings highlight that TNFα stimulates autophagy in osteoclasts in both experimental arthritis and RA. Of note, other proinflammatory cytokines, including interleukin (IL)-1 and IL-6, as well as TLR ligands, have also been shown to stimulate autophagy in other cell types,25–27 and may also contribute to the enhanced activation of autophagy in erosive joint disease. This may result in a highly stimulatory milieu to activate autophagy in inflamed RA joints. Indeed, treatment with glucocorticosteroids that inhibit the release of proinflammatory cytokines inhibits autophagy.28 ,29
TNFα has a prominent role in joint destruction in RA. Given its stimulatory effects on autophagy, we hypothesised that autophagy may regulate osteoclast differentiation and function. Indeed, we found that autophagy regulates the differentiation of monocytes into mature osteoclasts. Activation of autophagy by overexpression of Beclin1 enhanced the potential of monocytes to differentiate into mature osteoclasts with increased mRNA levels of NFATc1, OSCAR, TRAP and cathepsin K, and increased numbers and size of osteoclasts. By contrast, pharmacologic or genetic inhibition of autophagy reduced the expression of osteoclast markers, decreased the mean size of osteoclasts and reduced osteoclast counts in vitro and prevented synovial osteoclastogenesis in hTNFα tg mice in vivo. A recent study further highlighted that autophagy regulates hypoxia-induced osteoclastogenesis through HIF-1α signalling pathway.30 In addition to its role in osteoclast formation, we found that stimulation of autophagy enhanced the resorptive capacity of osteoclasts in vitro, whereas, inhibition of autophagy reduced bone resorption. These effects of autophagy on osteoclastic bone resorption may depend on Rab7. A very recent study demonstrated that autophagy-related proteins are essential to direct Rab7 to the ruffled border, regulate the fusion of secretory lysosomes, and release cathepsin K and other bone-degrading enzymes.18
In our study, we further demonstrate that inhibition of autophagy prevented joint destruction in experimental arthritis. Although clinical and histological signs of inflammation were comparable between Atg7fl/fl×LysMCre+ → hTNFα tg mice and controls, structural damage was strongly reduced in Atg7fl/fl×LysM Cre+→hTNFα tg mice. Both synovial osteoclastogenesis and bone erosions were decreased in Atg7fl/fl×LysMCre+→hTNFα tg mice. Of note, cartilage damage was also reduced in Atg7fl/fl×LysMCre+→hTNFα tg mice with decreased proteoglycan loss and chondrocyte apoptosis. Given the highly selective inhibition of autophagy in monocytic cells, the observed protection of cartilage in Atg7fl/fl×LysMCre+→hTNFα tg mice is most likely mediated by indirect effects. Indeed, the expression levels of the monocyte/macrophage derived cytokines IL-1β and IL-6 were both significantly decreased in serum of Atg7fl/fl×LysM Cre+→hTNFα tg mice compared with Atg7fl/fl×LysM Cre−→hTNFα tg mice. Inhibition of autophagy in infiltrating monocytes and resident macrophages may impair, and thus decrease fibroblast activation by impaired release of IL-1 and IL-6 to ameliorate cartilage destruction. However, further experiments are needed to address the underlying mechanism. These findings further highlight the key role of autophagy in TNFα-induced joint disease and suggest translational implications. Selective inhibitors are currently developed in the treatment of cancer patients, but are not available for clinical routine yet.31 ,32 Several drugs in clinical use, however, do have concomitant inhibitory effects on autophagy. Among those agents are chloroquine and hydroxychloroquine, which can prevent bone erosions and are established therapies of RA.33 ,34 Although less potent than bafilomycin A, chloroquine and hydroxychloroquine inhibit autophagy by raising the intralysosomal pH. This impairs protein degradation within the autophagosome and leads to the accumulation of ineffective autophagosomes.35 Although not exclusively mediated by inhibition of autophagy, the antiresorptive effects of both drugs, nevertheless, support the concept that inhibiting autophagy may be a promising therapeutic approach in the prevention of bone resorption in RA.
In summary, we provide evidence for a central role of autophagy in osteoclastogenesis and destructive joint disease. Autophagy is activated by TNFα in RA, and stimulates osteoclast differentiation. Selective inhibition of autophagy in monocytic cells prevents structural damage in hTNFα tg mice.
Acknowledgments
We thank Dr George Kollias (Alexander Fleming Biomedical Science Research Center, Vari, Greece) for providing the hTNFα tg mice. We thank Nadja Schröder-Kreß for technical help with the transmission electron microscopy. We also thank Madeleine Demleitner, Stefan Fritz, Maria Halter, Anna-Maria Herrmann and Verena Wäsch for excellent technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
Footnotes
Contributors Design of the study: NYL, GS, JHWD. Acquisition of data: NYL, CB, AG, TK, CS, SU, LEM, CD, AA. Interpretation of data: NYL, CB, AG, TK, CS, SU, LEM, AA, JHB, CB, GK, BS, OD, CD, JHWD. Manuscript preparation: NYL, CB, GS, JHWD.
Funding The study was supported by the Deutsche Forschungsgesellschaft (grants DI-1537/2-1, DI-1537/4-1, DI-1537/5-1, AK 144/1-1 and SCHE 1583/7-1), the Focus Programme SPP1468 (immunobone) of the Deutsche Forschungsgesellschaft and grant A40 of the Interdisciplinary Center of Clinical Research (IZKF) in Erlangen. The study was also supported by FP7 Masterswitch project of the European Union, the IMI funded project BTCure, and the carrier support award of the Ernst-Jung Foundation. N-YL holds a doctoral scholarship by Bavarian Research Foundation. Christian Beyer holds a research scholarship at the Interdisciplinary Center of Clinical Research (IZKF) in Erlangen.
Competing interests None.
Ethics approval Ethical Committee of the University of Erlangen-Nuremberg.
Provenance and peer review Not commissioned; externally peer reviewed.