


Abstract
The pathophysiology of cerebral ischemia involves multiple mechanisms including neuroinflammation mediated by activated microglia and infiltrating macrophages/monocytes. The present study employed a rat permanent middle cerebral artery occlusion (pMCAO) model to study effects of histone deacetylase (HDAC) inhibition on ischemia-induced brain infarction, neuroinflammation, gene expression, and neurological deficits. We found that post-pMCAO injections with HDAC inhibitors, valproic acid (VPA), sodium butyrate (SB), or trichostatin A (TSA), decreased brain infarct volume. Postinsult treatment with VPA or SB also suppressed microglial activation, reduced the number of microglia, and inhibited other inflammatory markers in the ischemic brain. The reduction in levels of acetylated histone H3 in the ischemic brain was prevented by treatment with VPA, SB, or TSA. Moreover, injections with HDAC inhibitors superinduced heat-shock protein 70 and blocked pMCAO-induced down-regulation of phospho-Akt, as well as ischemia-elicited up-regulation of p53, inducible nitric oxide synthase, and cyclooxygenase-2. The motor, sensory, and reflex performance of pMCAO rats was improved by VPA, SB, or TSA treatment. The beneficial effects of SB and VPA in reducing brain infarct volume and neurological deficits occurred when either drug was administrated at least 3 h after ischemic onset, and the behavioral improvement was long-lasting. Together, our results demonstrate robust neuroprotective effects of HDAC inhibitors against cerebral ischemia-induced brain injury. The neuroprotection probably involves multiple mechanisms including suppression of ischemia-induced cerebral inflammation. Given that there is no effective treatment for stroke, HDAC inhibitors, such as VPA, SB, and TSA, should be evaluated for their potential use for clinical trials in stroke patients.
Stroke, also referred to as cerebral ischemia, is a pathological condition resulting from occlusion or hemorrhage of blood vessels supplying oxygen and essential nutrients to the brain. In all cases, stroke ultimately induces death and/or dysfunction of brain cells, as well as neurological impairments that reflect the location and size of the ischemic brain area. Mechanisms leading to cell death during cerebral ischemic injury are complex, including excitotoxicity, ionic imbalance, oxidative/nitrosative stress, and inflammation (for review, see Lo et al., 2003). It is increasingly recognized that cerebral inflammation mediated by activated microglia and infiltrating leukocytes, including monocytes/macrophages, also plays a key role in focal ischemia-induced neurodegeneration. Activated microglia and invading leukocytes exert a cytotoxic function by releasing proinflammatory cytokine factors (IL-1, IL-2, and TNF-α), nitric oxide (NO), and reactive oxygen species, which contribute to brain infarction and excitotoxicity (Gregersen et al., 2000).
Valproic acid (2-propylpentanoic acid sodium salt; VPA), a drug commonly used to treat seizures and bipolar mood disorder, has been shown to have neuroprotective properties at therapeutic levels in cellular and animal models. In cultured neurons, VPA protects from glutamate-induced excitotoxicity (Kanai et al., 2004), thapsigargin-induced endoplasmic reticulum stress (Bown et al., 2000), and lipopolysaccharide (LPS)-induced dopaminergic neuronal death (Peng et al., 2005). VPA-induced neuroprotection against excitotoxicity is mimicked by inhibitors of histone deacetylase (HDAC) (Kanai et al., 2004), a recently identified target of VPA (Phiel et al., 2001). Postinsult treatment with VPA decreases brain infarct volume and neurological deficits in a transient ischemic model of rats subjected to middle cerebral artery occlusion (MCAO) (Ren et al., 2004). Like VPA, the short-chain fatty acid, sodium butyrate (butyric acid sodium salt; SB), and the hydroxamic acid trichostatin A ([R-(E,E)]-7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide; TSA) are HDAC inhibitors that suppress cell growth by cell cycle arrest and promote differentiation of normal and transformed cells (for review, see Drummond et al., 2005). SB and TSA have also been shown to have neuroprotective properties in neurons (for review, see Drummond et al., 2005; Langley et al., 2005; Leng and Chuang, 2006). These actions of VPA, SB, and TSA are probably related to HDAC inhibition, which leads to hyperacetylation of chromatin proteins and results in alteration in gene expression (for review, see Drummond et al., 2005).
Recently, VPA was shown to inhibit LPS-induced, microglia-mediated inflammation in midbrain neuron-glia cultures as demonstrated by inhibition of TNF-α secretion and NO production (Peng et al., 2005). Likewise, SB was reported to be anti-inflammatory in LPS-treated, brain-derived primary microglia but proinflammatory in transformed, proliferating N9 microglial cells (Huuskonen et al., 2004). The present study was undertaken to study whether postinsult treatment with HDAC inhibitors protects against brain injury and neurological deficits in a permanent MCAO (pMCAO) model of rats. We also attempted to investigate whether the HDAC inhibitor-induced neuroprotection is associated with suppression of pMCAO-induced activation of microglia/macrophage and to characterize HDAC inhibitor-elicited neuroprotective effects in the pMCAO model.
Materials and Methods
Focal Cerebral Ischemia. All experiments were approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of animals. Anesthesia of male Sprague-Dawley rats (240–260 g) was induced by 3% halothane (70% N2O/30% O2) and maintained with 1.5 to 3% halothane during surgery. Focal cerebral ischemia was performed as described previously (Ren et al., 2003, 2004), except that the reperfusion procedure was omitted. In brief, after incision of the neck dermis, the left common carotid artery was exposed, and the external carotid artery was ligated with a 4-0 suture. The internal carotid artery was isolated and separated from the vagus nerve. A 17-mm 4-0 nylon thread coated with silicon was inserted through a small puncture in the common carotid artery and fed into the left internal carotid artery and further advanced to the Circle of Willis to the origin of the left middle cerebral artery. The thread remained in position until the rats were sacrificed. At 20 min after MCAO, the cerebral blood flow in the ischemic brain area dropped to approximately 20% of the preischemic basal value (Ren et al., 2003). The body temperature was maintained at 36 to 37°C throughout the surgery with a heating pad. Sham-operated rats underwent identical surgery except that the thread was not inserted. Animals were divided into sham, vehicle, VPA-, SB-, or TSA-treated groups. The vehicle used for VPA and SB was normal saline, and for TSA, the vehicle was dimethyl sulfoxide (DMSO). The body temperature was found not to be significantly altered between the vehicle and VPA-treated ischemic rats measured at 20 min preischemia and various time (i.e., 20 min and 24 and 48 h) postischemia (data not shown).
Measurement of Infarct Volume. Animals were sacrificed by CO2 inhalation 24 h after pMCAO. Brains were rapidly removed and sectioned coronally at 2-mm thickness. The sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in saline for 20 min at 37°C and then fixed with 4% paraformaldehyde. The images of six continuous sections were captured with a digital camera and then analyzed for infarct volume with Adobe Photoshop 7.0 (Adobe Systems Inc., San Jose, CA). The infarct size was corrected for edema by subtracting the volume of normal tissue in the ischemic hemisphere from the volume of the corresponding area in the contralateral hemisphere, essentially as described previously (Ren et al., 2003). The infarction volume was calculated by summing the infarction areas of all sections and multiplying by the slice thickness.
Immunohistochemistry. One or 2 days after ischemia, rats were anesthetized and perfused transcardially with phosphate-buffered saline, pH 7.4. Excised brain tissues were rapidly immersed in dry ice-precooled isopentane and stored in a –80°C freezer. Twenty-micrometer coronal sections were cut onto glass slides using a cryostat. After fixation in 3.7% paraformaldehyde for 30 min at room temperature, endogenous peroxidase was inhibited by incubation with 0.3% H2O2 for 45 min. Nonspecific binding was blocked by incubating the sections in 5% nonfat dry milk solution for 45 min, as described previously (Ren et al., 2003). The sections were then incubated at 4°C overnight with antibodies against OX42 (polyclonal rat anti-mouse CD11b, 1:100 dilution; Serotec, Kidlington, Oxford, UK), ED1 (1:50; Chemicon International, Temecula, CA), or iNOS (1:1000; BD Biosciences, San Jose, CA). The bound antibody was detected using biotinylated goat-anti-mouse IgG-B (1:100; Chemicon International) in conjunction with horseradish peroxidase-conjugated streptavidin (1:500; Upstate Biotechnology, Lake Placid, NY). Between steps, the brain sections were rinsed three times in phosphate-buffered saline for 10 min and then visualized with 0.05% 3-3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO) and 8% nickel ammonium sulfate. Coronal sections were dehydrated, made translucent by immersion in xylene, and coverslipped. Negative control sections received identical treatment except that the primary antibody was omitted. OX42- and ED1-immunoreactive cells in coronal sections harvested at 24 and 48 h after pMCAO were quantified by a blind observer at 400× magnification under a microscope. In contrast, the numbers of OX42- and ED1-positive staining cells were quantified in the entire ischemic region from three sections per animal at different levels (anterior, bregma, +1.00 mm; middle, bregma, –3.14 mm; posterior, bregma, –4.80 mm). These levels of bregma were chosen because specific anatomical structures can be readily identified. For example, the lateral ventricle, hippocampus, and pineal recess of the third ventricle can be found at bregma +1.00, –3.14, and –4.80 mm, respectively.
Western Blotting Analysis. The ischemic brain hemispheres were homogenized in ice-cold lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 1 mM EGTA, 0.5 mM Na3VO4, 0.1% 2-mercaptoethanol, 1% Triton X-100, 50 mM NaF, 5 mM sodium pyrophosphate, 10 mM sodium β-glyceropyrophosphate, 0.1 mM phenylmethanesulfonyl fluoride, and protease inhibitor mixture) (Roche Diagnostics, Indianapolis, IN). The lysates were sonicated in ice for 45 s and centrifuged at 12,000 rpm for 20 min; the protein concentrations in the supernatant were determined using a bicinchoninic acid kit from Pierce (Rockford, IL). Twenty micrograms of protein in each well was subjected to SDS-polyacrylamide gel electrophoresis using a 4 to 12% Ready Gel (Invitrogen, Carlsbad, CA), followed by transfer of the protein bands to a polyvinylidene difluoride membrane. Primary antibodies against acetylated histone H3 (1:250; Upstate Biotechnology), HSP70 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Akt (Ser473) (1:200; Cell Signaling Technology, Danvers, MA), Bcl-2 (1:200; Santa Cruz Biotechnology), p53 (1:200; Santa Cruz Biotechnology), iNOS (1:200; Cell Signaling), and cyclooxygenase (COX)-2 (1:500; Santa Cruz Biotechnology) were incubated overnight at 4°C followed by incubation for 1 h with a horseradish peroxidase-conjugated secondary antibody at room temperature (1:1500; Santa Cruz Biotechnology). Protein bands were detected using an enhanced chemiluminescence system (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) and exposed to Hyperfilm (Amersham Biosciences). To serve as a protein loading control, the membrane was stripped and probed for β-actin using its specific antibody (1:5000; Sigma-Aldrich). The films were scanned by an optical densitometer to detect the intensity of protein bands and analyzed using UNSCAN-IT gel version 5.1 (Silk Scientific, Orem, UT).
Behavioral Studies. All behavioral tests were performed by three investigators who were blind to the experimental groups. Routinely, the tests were performed 1 day before pMCAO and 1 day after pMCAO, unless otherwise indicated. Before ischemic onset, all rats were able to perform the tests and did not exhibit asymmetries. After ischemia, epileptic rats were excluded from experiments.
Neurological Deficit Test. The score of neurological severity was a composite of motor, sensory, and reflex tests essentially as described previously (Li et al., 2000). Neurological severity scores included three tests where each animal was suspended by the tail (flexion of forelimb, head moved >10° to vertical axis, circling toward paralytic side), three tests in an open field (abnormal gait, circling walk toward paralytic side, and falling down), pinna reflex (a head shake when touching the auditory meatus), and visual placement test (stretching of forelimbs to meet an approaching object). Each test was scored as 1 for normal and 0 for abnormal, producing a summed severity of injury score graded on a scale of 0 to 8 with a maximal deficit score being 0 and normal being 8.
Rotorod Test. An accelerating Rotorod (4 to 40 rpm; San Diego Instrument, Inc., San Diego, CA) was used to assess the motor function of rats. The animals were trained for three trials 1 day before pMCAO. Each training trial was separated by 2.5 h. One day after surgery, rats underwent a single trial on the Rotorod.
Statistical Analysis. The data are expressed as mean ± S.E.M., and were analyzed using analysis of variance or Student's t test as appropriate. Differences with p ≤ 0.05 were considered statistically significant.
Results
Treatment with HDAC Inhibitors Reduces pMCAO-Induced Brain Infarct Volume. Three HDAC inhibitors, VPA, SB, and TSA, were examined for their effects on pMCAO-induced brain infarction, and their chemical structures are shown (Fig. 1, A, D, and G). To examine the neuroprotective effects of VPA against ischemia-induced brain damage, rats subjected to pMCAO were treated with 300 mg/kg VPA by s.c. injection every 12 h starting immediately after the onset of insult. Our previous study has shown that VPA at this dose inhibits HDAC activity, as shown by an increase in histone H3 acetylation in the intact contralateral brain hemisphere within 6 h after VPA injection, and exhibits neuroprotective effects on the ipsilateral hemisphere in a transient rat MCAO model (Ren et al., 2004). Brain damage was detected by TTC staining, and the infarct volume was calculated. pMCAO caused a cerebral infarct volume of approximately 300 mm3 in the ipsilateral brain hemisphere 24 h after ischemic onset, and postinsult VPA treatment decreased the infarct size by approximately 37% (Fig. 1, B and C). At this time, the plasma levels of VPA were 54.2 ± 3.5 μg/ml (n = 12), which is in the lower range of the therapeutic plasma levels used to treat bipolar disorder (50–125 μg/ml). Treatment with SB (300 mg/kg), another fatty acid HDAC inhibitor, by s.c. injection immediately after pMCAO followed by another injection 12 h later also decreased brain infarction determined at 24 h (Fig. 1E). The effect was dose-dependent with an approximately 50% decrease in infarct volume at the dose of 200 to 300 mg/kg, whereas the protective effect was less pronounced at higher doses, notably 700 mg/kg, which induced only a 22% decrease (Fig. 1F). These effective doses of SB were comparable with those used in mice by other investigators (Ferrante et al., 2003; Ryu et al., 2003). Likewise, postinsult injections with TSA (0.5 mg/kg s.c. immediately after pMCAO followed by another injection 12 h later), a potent hydroxamic acid HDAC inhibitor, decreased pMCAO-induced brain infarction by 34% (Fig. 1, H and I), compared with the vehicle (DMSO) control. The dose of TSA was also comparable with those used in mice and rats (Korzus et al., 2004; Leng and Chuang, 2006).

Postinsult treatment with HDAC inhibitors reduces brain infarct volume. Rats were subjected to pMCAO and injected with vehicle or indicated HDAC inhibitor immediately after the onset of ischemia, followed by another injection at 12 h. Rats were sacrificed at 24 h, and brain infarction in coronal sections was detected by TTC staining. A, chemical structure of VPA. B, TTC staining from a typical vehicle (normal saline)- or VPA (300 mg/kg s.c.)-treated rat. C, brain infarction volume was quantified from a group of vehicle-and VPA-treated animals. Data are mean ± S.E.M. from six rats in each group. D, chemical structure of SB. E, TTC staining from a typical vehicle (normal saline)- or SB (300 mg/kg s.c.)-treated rat. F, rats subjected to pMCAO were also injected with various doses of SB as indicated, and infarct volumes were determined in vehicle- or SB-treated animals. Data are mean ± S.E.M. from six rats in each group. G, chemical structure of TSA. H, TTC staining from a typical vehicle (DMSO)- or TSA (0.5 mg/kg s.c.)-treated rat. I, brain infarct volumes were quantified from a group of vehicle- or TSA-treated rats. Data are mean ± S.E.M. from a group of six rats in each group. ***, p < 0.001 compared with the respective vehicle-treated group.
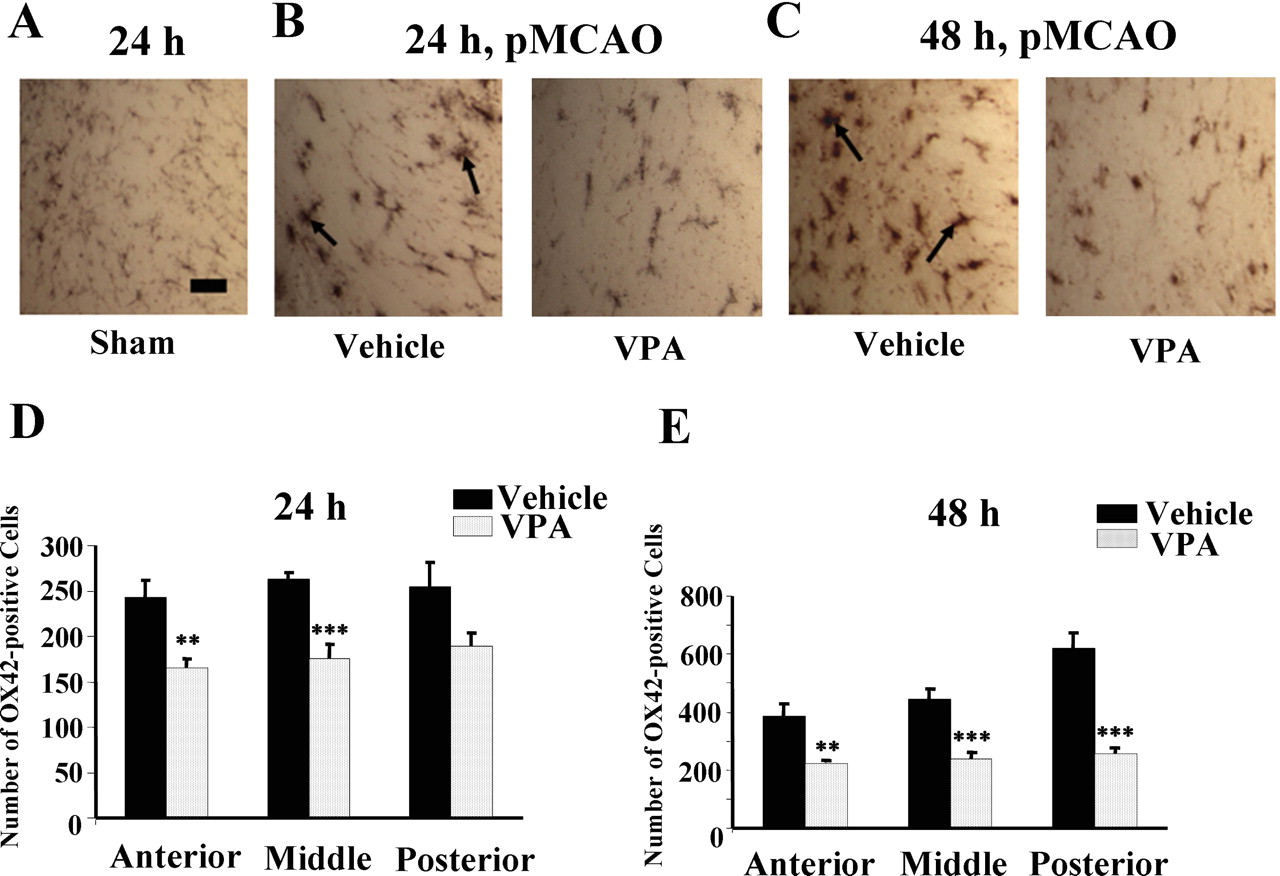
Treatment with VPA Exhibits Anti-Inflammatory Properties in the Ischemic Brain. We assessed effects of VPA treatment on microglia activation associated with pMCAO, which plays a critical role in ischemia-induced neurodegeneration (Gregersen et al., 2000). Microglia express complement type 3 receptors (CD11b/CD18 complex), which are recognized by the monoclonal antibody against OX-42 in the rat (Stoll et al., 1998). OX-42 immunostaining of microglia revealed the presence of activated microglia characterized by larger, amoeboid morphology in the ischemic corpus callosum 24 and 48 h after pMCAO (Fig. 2, A–C). Postinsult VPA administration suppressed pMCAO-induced microglia activation in the corresponding brain area of treated rats at both time points. The numbers of OX42-positive microglia at the anterior, middle, and posterior sections of the ipsilateral brain hemisphere were all significantly reduced by VPA treatment at both 24 and 48 h (Fig. 2, D and E). We also examined the effects of VPA on the number of cells stained positively by an antibody to ED1, a marker for monocytes/macrophages and, to some extent, for activated microglia (Stoll et al., 1998). Monocytes/macrophages contribute to neuroinflammation by infiltrating into the damaged brain area due to disruption of the blood-brain barrier after cerebral stroke (Gregersen et al., 2000). ED1-positive cells, characterized by a larger round shape, were detected in the ischemic frontal cortex, and this ED1 immunostaining determined 24 and 48 h after pMCAO was suppressed by VPA treatment in the corresponding brain area (Fig. 3, A and B). Quantified results showed that the numbers of ED1-positive cells in the anterior, middle, and posterior sections of the ipsilateral hemisphere were all markedly decreased by VPA treatment determined at both time points (Fig. 3, C and D).
It has been reported that iNOS is induced in the brain after ischemic stroke (Nogawa et al., 1998). Immunohistochemical staining of iNOS confirmed that its immunoreactivity was undetectable in the striatum of the contralateral hemisphere but abundantly present in cells as well as vascular structures in the ischemic striatum 24 h after pMCAO (Fig. 4, A, C, and E). VPA treatment essentially depleted iNOS immunoreactivity in the ischemic striatum (Fig. 4, D and F). Similar inhibitory effects of VPA on iNOS immunoreactivity were found in the ischemic frontal cortex (Fig. 4, G–J).

Postinsult VPA treatment reduces the number of OX42-positive microglia in the ipsilateral brain hemisphere after pMCAO. Rats were subjected to pMCAO and postinsult injection with vehicle or VPA every 12 h as described in the legend to Fig. 1. Animals were sacrificed at 24 h or 48 h. A, immunostaining of microglia with anti-OX42-antibody in the corpus callosum area of a coronal section of a sham-operated rat. The OX42-positive cells showed small cell bodies with ramified, thin processes characteristic of resting microglia. Bar, 30 μm. B and C, OX42-positive microglia in the corresponding ischemic corpus callosum determined 24 and 48 h after pMCAO, respectively. Arrows in vehicle-treated sample, activated microglia with amoeboid morphology. D and E, number of OX42-positive cells determined at 24 and 48 h, respectively, in the entire anterior, middle, and posterior sections of the ipsilateral hemisphere. Data are mean ± S.E.M. from six animals in each group. **, p < 0.01; ***, p < 0.001 compared with the corresponding vehicle group.
VPA and TSA Regulate Levels of Proteins Related to Apoptosis. Western blotting was performed to assess changes in levels of cytoprotective and apoptotic proteins. The levels of acetylated histone H3 were decreased 24 h after pMCAO in the ipsilateral brain hemisphere, and this reduction was blocked by postinsult VPA injections (Fig. 5, A and B), suggesting VPA-induced inhibition of HDAC activity.
The levels of cytoprotective HSP70 in the ischemic, ipsilateral hemisphere were increased by pMCAO compared with the sham-operated control, and this increase was potentiated by VPA treatment (Fig. 5, A and C). The levels of p53, a transcription factor and proapoptotic protein negatively regulated by HDACs (Juan et al., 2000), were also markedly increased in the ischemic hemisphere compared with the sham group, and this p53 up-regulation was suppressed by VPA treatment (Fig. 5, A and D). The protein levels of β-actin used as loading control were unchanged under these experimental conditions.
Similar to VPA, TSA treatment blocked the loss of acetylated histone H3 and potentiated HSP70 induction but suppressed p53 up-regulation in the ischemic brain hemisphere (Fig. 5E). In addition, levels of two cytoprotective factors, Bcl-2 and phospho-Akt, were decreased after pMCAO in the ischemic brain, compared with the sham-operated control, and these effects were inhibited by TSA injections.
SB Also Shows Anti-Inflammatory and Antiapoptotic Effects in Ischemic Rats. Similar to the effects of VPA, SB treatment suppressed the activation and number of microglia detected by OX42-immunostaining in the infarcted corpus collosum (Fig. 6A). Quantification of OX42-positive cells revealed that their numbers were decreased by almost 30% in the anterior, middle, and posterior sections of the ischemic hemisphere (Fig. 6B). Western blots showed that pMCAO-induced reduction of acetylated histone H3 levels was prevented by postinsult SB treatment (Fig. 6, A and B), again suggesting an inhibition of HDAC activity. The up-regulation of HSP70 protein levels was robustly enhanced by SB treatment (Fig. 7, A and C), whereas the loss of levels of phospho-Akt was prevented by SB treatment (Fig. 7, A and D). The pMCAO-induced up-regulation of p53 and iNOS was also blocked by SB (Fig. 7, A and E). Furthermore, pMCAO-induced increase of COX-2 protein, the rate-limiting enzyme for prostanoid synthesis and an inflammatory marker, was robustly suppressed by SB treatment (Fig. 7, A and F).

Postinsult VPA treatment reduces the number of ED1 (a preferential monocyte/macrophage marker)-positive cells in the ipsilateral brain hemisphere after pMCAO. Experimental conditions are as described in the legend to Fig. 2. A and B, representative immunostaining of ED1-positive cells in the ischemic frontal cortex determined 24 and 48 h after pMCAO, respectively. Bar, 20 μm. Note that the morphology of ED1-positive cells in the ischemic area was bigger and more rounded and differed from that of activated microglia. C and D, number of ED1-positive cells determined at 24 and 48 h, respectively, in the entire anterior, middle, or posterior sections of the ipsilateral hemisphere. Data are mean ± S.E.M. from six rats in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the corresponding vehicle group.
Treatment with SB, VPA, or TSA Improves Neurological Performance in Ischemic Rats. Behavioral assessments of motor, sensory, and reflex functions were performed in pMCAO and sham-operated rats. In vehicle-treated pMCAO rats, the neurological scores determined by an eight-point test were decreased by approximately 70% at 24 h, whereas the sham-operated group showed no significant neurological impairment (Fig. 8A). Markedly, postinsult treatment with SB or VPA significantly improved neurological performance.
Postinsult treatment with TSA also increased the neurological activity measured in the eight-point test (Fig. 8B). The long-term effects of postinsult SB treatment on functional outcome were further assessed by the eight-point behavioral test. There was a significant increase in the neurological performance score with time after pMCAO in the saline-treated group (p < 0.05 from days 2–7 or 2–11), and daily SB treatment significantly improved upon this neurological function at 2, 4, 7, and 11 days after ischemia (Fig. 8C). The neurological functions seemed to be normal at any of these time points in the sham-operated group.
The Beneficial Time Window for VPA and SB Is at Least 3 h after Ischemic Onset. We next examined whether beneficial effects of VPA and SB were observed by delayed administration of either drug. When VPA (300 mg/kg) was first injected 3 h after pMCAO, a significant decrease (by 32.7%) in infarct volume determined at 24 h was observed (Fig. 9A). This protective effect in the pMCAO rats was accompanied by a significant increase in the neurological scores on the eight-point test (Fig. 9B). The Rotorod test has been validated as a means of assessing motor coordination in rats, and impaired Rotorod performance is significantly correlated with increasing brain infarct volume (Rogers et al., 1997). Our results showed that the retention time on the accelerating Rotorod of VPA-treated rats was increased from 20 to almost 50 s (Fig. 9C). Similar effects on the reduction of brain infarction and improvement of neurological performance were observed in pMCAO rats first injected with SB (300 mg/kg) 3 h after the ischemic onset (Fig. 10, A–C). When SB was first injected 6 h after pMCAO, a significant reduction (by 33.1%) in infarction was observed (Fig. 11A). The behavioral results of the eight-point neurological and Rotorod tests did show a tendency toward improvement; however, these differences did not reach statistical significance (Fig. 11, B and C).
Discussion
The present study employed a rat pMCAO model to investigate the neuroprotective effects of VPA, SB, and TSA. Compared with transient MCAO, pMCAO produces a more severe and rapid brain infarction with a smaller and more short-lived penumbra. The process of pMCAO also favors the mechanisms of necrotic-type cell death, presumably due to rapid loss of glucose utilization and protein synthesis following permanent ischemia (for review, see Lo et al., 2003). Despite the severity of brain pathology induced by pMCAO, we found that postinsult treatment with VPA, SB, or TSA effectively reduced brain infarction and suppressed neurological deficits (Figs. 1 and 8). The dose of VPA (300 mg/kg s.c. twice daily) produced a level of 54 μg/ml (or 0.33 mM) in the plasma. The plasma levels of SB and TSA were not determined in our experimental conditions. However, based on the assumption that the pharmacokinetic parameters of both drugs are similar to those of VPA, the doses of SB (300 mg/kg s.c. twice daily) and TSA (0.5 mg/kg s.c. twice daily) can be projected to have estimated plasma levels of 54 (or approximately 0.5 mM) and 0.9 μg/ml (or approximately 3 μM), respectively.
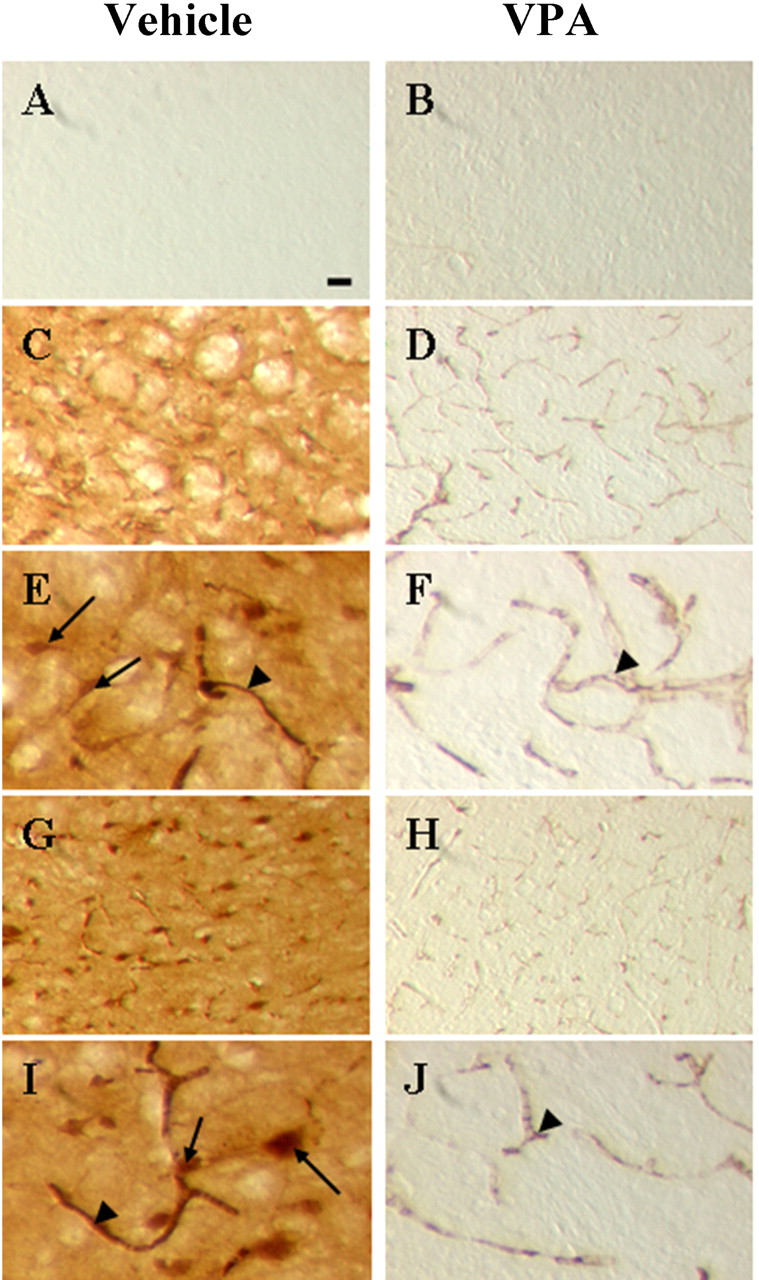
pMCAO-induced iNOS immunohistochemical staining was blocked by postinsult VPA treatment. Experimental conditions are as described in the legend to Fig. 1B. Immunohistochemistry of iNOS-positive cells was examined in the striatum and frontal cortex of the ipsilateral hemisphere. A and B, lack of iNOS-positive cells in the striatum of the contralateral hemisphere of vehicle- and VPA-treated rats, respectively (magnification, ×40). C and D, iNOS-positive cells in the corresponding area of the striatum of the ipsilateral hemisphere of vehicle- and VPA-treated rats, respectively (magnification, ×40). E and F, same as C and D except that the magnification was ×100. Arrow, iNOS-positive cell; arrowhead, iNOS-positive vessel. G and H, iNOS-positive cells in the corresponding area of the frontal cortex of the ipsilateral hemisphere of vehicle- and VPA-treated animals, respectively (magnification, ×40). I and J, same as G and H except that the magnification was ×100. Bar, 30 μm.
Importantly, the beneficial effects were evident when VPA or SB was administrated at least 3 h after the ischemic onset (Figs. 9, 10, 11). This time window, together with the long-lasting neurological improvement (Fig. 8), suggests that HDAC inhibitors might have utility in treating acute stroke.

Effects of postinsult VPA or TSA treatment on levels of Ac-H3 and antiapoptotic/proapoptotic proteins in the ischemic brain hemisphere of pMCAO rats. Experimental conditions are as described in the legend to Fig. 1. Animals were sacrificed 24 h after pMCAO, and the lysates of the ipsilateral hemisphere were prepared for Western blotting analysis. A, Western blots of acetylated histone H3 (Ac-H3), HSP70, p53, and β-actin from sham-operated rats and pMCAO rats treated with vehicle or VPA. B to D, quantified results of Ac-H3, HSP70, and p53, respectively. Data are mean ± S.E.M. of percentage of sham-operated control with three to four animals in each group. *, p < 0.05; **, p < 0.01 between the indicated groups. E, Western blots of Ac-H3, HSP70, p53, Bcl-2, p-Akt, and β-actin from sham-operated and pMCAO rats treated with vehicle (DMSO) or TSA. Results shown are from a representative experiment that included three to four rats in each group.
One striking feature of our results is that the neuroprotective effects of VPA and SB are associated with inhibition of pMCAO-induced neuroinflammation. It is now generally accepted that neuroinflammation mediated by microglia and/or monocyte/macrophages has a prominent role in ischemia-induced brain injury. Inflammatory cytokines (TNF-α and IL-1β), iNOS and COX-2 are overexpressed to induce the inflammatory response. After stroke, many of the leukocytes including monocytes and macrophages move from the blood vessels and accumulate in the infarct zone to contribute to neuroinflammation and neurodegeneration (for review, see Zheng and Yenari, 2004). Our results showed that VPA treatment significantly reduced the number of both OX42-positive microglia and ED1-positive monocytes/macrophages following pMCAO (Figs. 2 and 3). A similar decrease in the number of microglia in the ischemic brain was also observed in pMCAO rats treated with SB (Fig. 6). In rat midbrain microglia-enriched cultures, treatment with VPA, SB, or TSA has been shown to inhibit LPS-induced microglial activation, which is associated with a decrease in microglial number (Peng et al., 2005; Chen et al., 2006) Our preliminary data showed that postinsult VPA treatment induced a marked increase in the population of microglia that was stained positive for caspase-3 in the ischemic cortex, consistent with the notion that HDAC inhibition induces apoptosis of microglia. Thus, HDAC inhibitor-induced apoptosis of microglia and monocytes/macrophages probably contributes to antineuroinflammation in the ischemic brain of pMCAO rats. On the other hand, it has been suggested that VPA may suppress LPS-induced production of inflammatory cytokines via inhibition of NF-κB transcriptional activity (Ichiyama et al., 2000).

Postinsult SB treatment reduces the number of OX42-positive microglia in the ipsilateral brain hemisphere after pMCAO. Rats were subjected to pMCAO and postinsult injection with vehicle or SB (300 mg/kg), as described in the legend to Fig. 1E. Animals were sacrificed 24 h after the onset of pMCAO. A, immunostaining of OX42-positive microglia in the corresponding area of the ischemic corpus callosum of a coronal section from vehicle- and SB-treated rats. Bar, 30 μm. B, number of OX42-positive cells in the anterior, middle, and posterior sections of the ipsilateral hemisphere. Data are mean ± S.E.M. from six animals in each group. ***, p < 0.001 compared with the corresponding vehicle group.
In midbrain neuron-glia cocultures, VPA treatment robustly suppresses LPS-induced secretion of proinflammatory TNF-α and production of NO (Peng et al., 2005). The present study showed that the expression levels of iNOS in the ischemic brain of pMCAO rats were blocked by VPA or SB treatment (Figs. 4 and 7). The role of iNOS in ischemia-induced pathology is supported by the observation that iNOS gene knockout attenuates ischemia-induced brain damage and neurological deficits (Iadecola et al., 1997). pMCAO-induced COX-2 overexpression was blocked by postinsult treatment with SB when examined 24 h after the onset of ischemia (Fig. 7). In rodents and humans, cerebral ischemia causes COX-2 overexpression in neurons, glia, vascular cells, and inflammatory cells that invade the ischemic area (Iadecola and Gorelick, 2005). COX-2 catalyzes the production of prostanoids and free radicals, both of which contribute to the deleterious effects of neuroinflammation and excitotoxicity resulting from cerebral ischemia. It has also been suggested that NO, produced by iNOS after focal cerebral ischemia, regulates the activity of COX-2 (Nogawa et al., 1998), possibly via nitrosylation and activation of this enzyme (Kim et al., 2005). Moreover, inhibition of COX-2 leads to attenuation of ischemic injury (Sugimoto and Iadecola, 2003), whereas transgenic COX-2 overexpression exacerbates cerebral infarction (Doré et al., 2003). Collectively, our results strongly suggest that anti-inflammation induced by HDAC inhibitors contributes to neuroprotection against cerebral ischemia, rather than being the result of neuroprotection induced by these drugs.

Effects of postinsult SB treatment on protein levels of acetylated histone H3 (Ac-H3), HSP70, p-Akt (Ser473), p53, iNOS, and COX-2 in the ischemic brain hemisphere of pMCAO rats. Experimental conditions are as described in the legend to Fig. 1E. The dose of SB was 300 mg/kg, and the animals were sacrificed 24 h after pMCAO followed by preparation of lysates from the ipsilateral hemisphere. A, Western blots of Ac-H3, HSP70, p-Akt, p53, iNOS, COX-2, and β-actin from sham-operated rats and pMCAO rats treated with vehicle or VPA. B to F, quantified results of Ac-H3, HSP70, p-Akt, p53, and COX-2, respectively. Data are mean ± S.E.M. of percentage of sham-operated control with three to four animals in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the vehicle group. †††, p < 0.001 compared with the sham group in C.

Postinsult treatment with VPA, SB, or TSA improves neurological performance of pMCAO rats. A, rats were subjected to pMCAO followed by postinsult injections with VPA (300 mg/kg) or SB (300 mg/kg), as described in the legend to Fig. 1, B and E. Eight independent behavioral tests were conducted 24 h after pMCAO to evaluate motor, sensory, and reflux functions. A total score of eight reflects normal neurological performance. Data are mean ± S.E.M. of the scores from sham-operated and pMCAO rats treated with vehicle, SB, or VPA (n = 11 in each group). *, p < 0.05; **, p < 0.01; ***, p < 0.001 between the indicated groups. B, sham-operated or pMCAO rats were injected with vehicle (DMSO) or TSA (0.5 mg/kg s.c.) immediately after the ischemic onset and followed by another injection 12 h later. At 24 h, eight-point neurological tests were performed. Data are mean ± S.E.M. of the scores from sham-operated (n = 10) and pMCAO rats injected with vehicle or TSA (n = 8 in each group). ***, p < 0.001 between the indicated groups. C, rats were subjected to pMCAO and injected with SB (300 mg/kg) immediately after the ischemic onset followed by s.c. injections every 12 h for the first 2 days and thereafter followed by daily injections. The eight-point behavioral tests were conducted at the indicated time points. The numbers of shamoperated, pMCAO-vehicle treated, and pMCAO-SB treated rats were 8, 9, and 11, respectively. Data are mean ± S.E.M. of the scores in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 between the indicated groups.

VPA injected 3 h after ischemia still induces a reduction in brain infarction and improvement of neurological performance. Experimental conditions are as described in the legend to Fig. 1B, except that VPA or vehicle was first injected 3 h after the ischemic onset. At 24 h after pMCAO, animals were assessed for neurological performance by the eight-point behavioral test and accelerating Rotorod test. Animals were then immediately sacrificed for the determination of brain infarct volume. A, TTC staining of brain infarction in six coronal sections was performed and the quantified results of the infarct volume are shown. Data are mean ± S.E.M. from six rats in each group. B, quantified results of the eight-point behavioral test for neurological performance. Data are mean ± S.E.M. of the performance score (n = 12 in the vehicle group; n = 14 in the VPA-treated group). C, quantified results of the Rotorod test. Data are mean ± S.E.M. of the retention time on the accelerating Rotorod (n = 11 in sham-operated group, n = 12 in the pMCAO-vehicle group, n = 14 in pMCAO-VPA group). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the respective vehicle group. †, p < 0.05 compared with the sham group in C.
It is becoming clear that chromatin structural remodeling via changes in levels of histone acetylation is a key mechanism underlying the regulation of gene expression (for review, see Drummond et al., 2005). Acetylation of histones is catalyzed by histone acetyltransferase, whereas their deacetylation is mediated by HDACs. Following pMCAO, there was a marked decrease in the acetylation levels of histone H3 in the ischemic brain (Figs. 5 and 7), similar to results from our previous study using a rat transient MCAO model (Ren et al., 2004). This decrease in acetylated histone could be related to inhibition of histone acetyltransferase and/or activation of HDAC following ischemic insult. In any event, MCAO-induced loss of histone acetylation was prevented by postinsult treatment with VPA, SB, or TSA (Figs. 5 and 7). Together, these results suggest that HDAC activity is inhibited in the rat brain by treatment with these three drugs under our experimental conditions. VPA is a short-chain fatty acid structurally similar to SB and has been shown to inhibit multiple HDAC subtypes from classes I and II (but not HDAC6 and HDAC10) (Gurvich et al., 2004), whereas TSA is structurally dissimilar (Fig. 1G) and has less selectivity for HDAC subtypes. The subtype of HDAC involved in HDAC inhibitor-induced neuroprotection against cerebral infarction awaits identification.

SB injected 3 h after ischemia still shows a reduction in brain infarction and improvement of neurological performance. Experimental conditions are as described in the legend to Fig. 1E, except that SB or vehicle was first injected 3 h after ischemic onset. At 24 h after pMCAO, animal were assessed for neurological performance by the eight-point behavioral test and accelerating Rotorod test. Animals were then immediately sacrificed for the determination of brain infarct volume. A, TTC staining of brain infarction in six coronal sections was performed, and the quantified results of the infarct volume are shown. Data are mean ± S.E.M. from vehicle (n = 6) and SB-treated (n = 11) rats. B, quantified results of the eight-point behavioral test for neurological performance. Data are mean ± S.E.M. of the performance score (n = 6 in the vehicle group; n = 10 in the SB-treated group). C, quantified results of the Rotorod test. Data are mean ± S.E.M. of the retention time on the accelerating Rotorod (n = 11 in sham-operated group as shown Fig. 9D; n = 6 in pMCAO-vehicle group; n = 11 in pMCAO-SB group). *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with the respective vehicle group.
The superinduction of HSP70 in ischemic brain by postinsult VPA, SB, or TSA treatment is possibly due to HDAC inhibition, which results in altered chromatin to a more open conformation, which then favors HSP70 transcriptional initiation. This notion is supported by our observation that HSP70 levels are also increased by VPA treatment in normal rats (Ren et al., 2004). VPA, SB, and TSA have also been shown to induce HSP70 in Xenopus and Drosophila (for review, see Langley et al., 2005). In addition to being a molecular chaperone assisting in proper protein folding, HSP70 has several critical effects against apoptotic and necrotic cell death. Overexpression of HSP70 in mice has been shown to provide protection from cerebral ischemia in an animal model of stroke (Rajdev et al., 2000; Hoehn et al., 2001). HSP70 up-regulation also leads to increased expression of antiapoptotic Bcl-2 and suppresses microglia/monocyte activation following experimental stroke (Yenari et al., 2005). HSP70 and Bcl-2 were recently reported to be increased in the brain of ischemic mice treated with another HDAC inhibitor, suberoylanilide hydroxamic acid, which also displayed neuroprotective effects (Faraco et al., 2006). The levels of phospho-Akt, which has suggested involvement in the pathophysiology of and neuroprotection against cerebral ischemia (Zhao et al., 2005), were decreased by pMCAO, and this reduction was blocked by TSA or SB treatment (Figs. 5 and 7). Moreover, the down-regulation of cytoprotective Bcl-2 in the ischemic brain was prevented by TSA injections (Fig. 5E).

Effects of SB injected 6 h postinsult on pMCAO-induced brain infarction and neurological deficits. Experimental conditions are as described in the legend to Fig. 1E, except that SB or vehicle was first injected 6 h after ischemic onset. A, TTC staining of brain infarction in six coronal sections was performed, and the quantified results of the infarct volume are shown. Data are mean ± S.E.M. from six rats in each group. ***, p < 0.001 compared with the vehicle group. B, quantified results of the eight-point behavioral test. Data are mean ± S.E.M. of performance score (n = 8 in the vehicle group; n = 8 in the SB-treated group; p = 0.1633 between groups). C, quantified results of the Rotorod test. Data are mean ± S.E.M. of the retention time on Rotorod (n = 9 in the vehicle group; n = 8 in the SB-treated group; p = 0.2268 between groups).
HDAC inhibitor-induced neuroprotection in the ischemic brain also seems to involve changes in the expression of proapoptotic proteins. For example, p53, which is negatively regulated by HDACs (Juan et al., 2000), was also up-regulated by pMCAO (Figs. 5 and 7), confirming results of previous reports (Watanabe et al., 1999), and the p53 up-regulation was blocked by postinsult VPA, SB, or TSA treatment (Figs. 5 and 7). The role of p53 in cerebral ischemia is suggested by the observation that pifithrin α, an inhibitor of p53 binding to its DNA sites, reduces the expression of the p53 downstream gene, p21WAF, and decreases the number of apoptotic cells in the ischemic brain (Leker et al., 2004). Thus, the inhibition of p53 protein levels by VPA, SB, and TSA probably contributes to the neuroprotective effects of both HDAC inhibitors against ischemic brain damage.
It should be noted that VPA, SB, and/or TSA also have other neuroprotective and neurotrophic actions. These include their ability to acetylate the neuroprotective transcription factor, Sp-1 (Ryu et al., 2003), and to induce neuroprotective proteins such as Bcl-2 (Chen et al., 1999), and glucose-regulated protein 78 (Bown et al., 2000). Conversely, these drugs inhibit the excitotoxicity-induced nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase (Kanai et al., 2004), which has been shown to have a proapoptotic role in neurons (for review, see Chuang et al., 2005). HDAC inhibitors also induce brain-derived neurotrophic factor in rat brain neurons (Fukumoto et al., 2001) and brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in cultured astrocytes from rat midbrain, causing robust neurite outgrowth (Chen et al., 2006). Collectively, our present results together with these reports suggest that HDAC inhibitor-induced neuroprotection following ischemia probably involves multiple mechanisms of action, including their ability to markedly inhibit ischemia-induced cerebral inflammation.
Acknowledgments
We thank Charlotte Wiest for assistance in behavioral tests and Peter Leeds of National Institute of Mental Health and the National Institutes of Health Fellows Editorial Board for editorial assistance.
Footnotes
-
This study was supported by the Intramural Research Program Research Fund of the National Institute of Mental Health, National Institutes of Health.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.107.120188.
-
ABBREVIATIONS: IL, interleukin; TNF, tumor necrosis factor; NO, nitric oxide; VPA, valproic acid (2-propylpentanoic acid sodium salt); LPS, lipopolysaccharide; HDAC, histone deacetylase; MCAO, middle cerebral artery occlusion; SB, sodium butyrate (butyric acid sodium salt); TSA, trichostatin A ([R-(E,E)]-7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide); pMCAO, permanent middle cerebral artery occlusion; DMSO, dimethyl sulfoxide; TTC, 2,3,5,-triphenyltetrazolium chloride; iNOS, inducible nitric-oxide synthase; HSP, heat shock protein; COX, cyclooxygenase.
- Received January 19, 2007.
- Accepted March 16, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}