Article Text

Abstract
Objective The objective of this study was to comparatively analyse the gastric fluid (GF) microbiota between patients with functional dyspepsia (FD) and healthy controls (HC), and to assess the effect of probiotics on the microbiota.
Design Twenty-four Japanese patients with FD who met the Rome III definition and 21 age-matched and gender-matched HC volunteers were enrolled. The patients with FD had been treated with LG21, a probiotic strain. The GF was sampled after an overnight fast using a nasogastric tube. The bile acids concentration was determined by ELISA. The V3-V4 region of 16S rRNA gene was amplified using bacterial DNA from the GF, and then about 30 000 high-quality amplicons per sample were grouped into operational taxonomic units for analyses.
Results The ratio of GF samples in which the bile acids were detectable was significantly greater in the FD than in the HC groups. In the bacterial composition analysis at the phylum level, the GF microbiota had a Bacteroidetes > Proteobacteria abundance and an absence of Acidobacteria in the FD group, in contrast, the GF microbiota had a Bacteroidetes < Proteobacteria abundance and the presence of Acidobacteria in the HC group. Probiotic therapy in patients with FD shifted the composition of the GF microbiota to that observed in the HC volunteers.
Conclusions Alteration in the GF microbiota was found in patients with FD compared with HC volunteers. Reflux of the small intestinal contents, including bile acid and intestinal bacteria, to the stomach was suggested to induce a bacterial composition change and be involved in the pathophysiology underlying FD. Probiotics appear effective in the treatment of FD through the normalisation of gastric microbiota.
Trial registration number UMINCTR 000022026; Results.
- BACTERIAL PATHOGENESIS
- FUNCTIONAL DYSPEPSIA
- PROBIOTICS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Summary box
What is already known about this subject?
▸ Little is known about the gastric microbiota or its role in the pathophysiology of functional dyspepsia (FD).
What are the new findings?
▸ Alteration in the gastric fluid microbiota characterised by Bacteroidetes > Proteobacteria abundance and the absence of Acidobacteria at the phylum level was found in the gastric fluid of patients with FD. Probiotic therapy in patients with FD shifted the composition of the gastric fluid microbiota to that observed in the healthy controls. Reflux of small intestinal contents including bile acids and intestinal bacteria to the stomach was suggested to induce such bacterial composition change and be involved in the pathophysiology underlying FD.
How might it impact on clinical practice in the foreseeable future?
▸ The finding of an FD-type phylum profile can be used to characterise patients with FD and may serve as an objective biomarker for both the diagnosis and treatment of FD.
Introduction
Functional dyspepsia (FD) is a clinical problem of considerable magnitude for the healthcare because of the high prevalence and recurrent nature of symptoms, which consist of postprandial distress and/or epigastric pain syndrome that are unexplained after a routine clinical evaluation.1 While the exact pathophysiology of FD remains to be clarified, gastric motility disturbance, such as delayed gastric emptying and impaired gastric accommodation after meals, and visceral hypersensitivity have been postulated as critical underlying mechanisms. Although there is much evidence to suggest that dysbiosis of the gut microbiota is involved in the pathophysiology of irritable bowel syndrome (IBS), a functional gastrointestinal disorder (FGID) originating from the intestine, little is known about the gastric microbiota or their role in the pathophysiology of FD, an FGID originating from the stomach and possibly the duodenum.
As antibiotic therapy targeting Helicobacter pylori appears to be effective in treating the FD symptoms in some patients, the therapeutic effects on the symptoms may not be simply mediated by the eradication of H. pylori but by the involvement of other antibacterial effects.2 Indeed, in our previous study examining the therapeutic effect of the probiotic strain Lactobacillus gasseri OLL2716 (LG21) on FD associated with H. pylori infection, the severity of postprandial distress syndrome was significantly lower after LG21 treatment than before the treatment, whereas no significant difference was found in the laboratory tests indicating the number and activity of H. pylori in the stomach, such as the H. pylori stool antigen and urea breath tests.3 As the normalisation of the perturbed microbiota is one of the dominant mechanisms underlying the effects of probiotics,4 such improvement in the FD symptoms in patients infected with H. pylori might be associated with a change in the gastric indigenous bacteria other than H. pylori.
Recently, Nakae et al5 reported that the overall bacterial community structure in the gastric fluid (GF) of patients with FD was significantly different from that in the GF of healthy control (HC) subjects when examined using the terminal restriction fragment polymorphism (T-RFLP) method. Furthermore, this bacteriological change was restored by treatment with LG21. However, the T-RFLP is a limited method and is therefore unable to obtain comprehensive information to fully elucidate the composition of microbiota. Therefore, in the present study, we used high-throughput bacterial 16S rRNA gene sequencing and bioinformatics technology to conduct analyses of the difference in the GF microbiota between FD and HC subjects, and the effect of probiotics on the GF microbiota in the patients with FD. As a result, we obtained definitive evidence that an alteration occurred in the GF microbiota of patients with FD, and the bacterial composition was shifted to that observed in the HC subjects by LG21 yogurt treatment.
Methods
Participants
Twenty-four Japanese patients with FD who met the definition of the Rome III classification were enrolled (table 1). The exclusion criteria were the use of antimicrobials within the previous 3 months, the use of acid-suppressive drugs, serological positivity for anti-H. pylori antibodies and a history of gastrointestinal (GI) or hepatobiliary surgery. A total of 21 healthy volunteers were also enrolled as HCs. The exclusion criteria were the same as those for the patients with FD. The ethics committee of Tokai University Hospital approved the study (13R-324), and written informed consent was obtained from all of the participants. A clinical part of this study is registered on UMIN-CTR website (http://www.umin.ac.jp/ctr; Test 000022026).
Demographic and laboratory data
Study protocol
All of the patients with FD were asked to consume 118 g of yogurt containing 109 colony-forming units (CFUs) of LG21 (LG21 yogurt) every day for a 12-week treatment period. The patients were informed that the end point of the study was to examine the influence of the yogurt on ‘the gastric fluid microbiota’. The compliance (days of having consumed the yogurt) was evaluated through diaries written by the subjects. The patients underwent examinations twice—before and after the yogurt treatment. In the morning on the day of examination, GF was sampled from the subjects after an overnight fast. For the sampling of GF, a nasogastric tube was inserted into the stomach through the nostril and then as much GF as possible was aspirated using a disposable syringe connected to the tube and transferred into a test tube. The participants were then asked to fill out a frequency scale for symptoms of gastroesophageal reflux disease (FSSG) questionnaire.6 This questionnaire is composed of 12 questions. Seven questions are related to gastro-oesophageal reflux or epigastric pain-like, and five are related to dysmotility-like dyspepsia or postprandial distress-like symptoms.
Laboratory examinations
The pH value of GF was measured using a pH meter (M-7; Horiba, Tokyo, Japan). To count the number of anaerobic and aerobic bacteria using culturing methods, Luria-Bertani (LB) and Tryptic soy (TS) agar plates, respectively, were used. The sum of the CFUs on both agar plates was used as the bacterial count. To measure the concentration of bile acids in the GF, an EFBA-100 Kit (BioAssay Systems, Hayward, California, USA) was used in accordance with the manufacturer's instruction. This assay kit can detect 12 different types of bile acids typically found in mammals including cholic and chenodeoxycholic acids. The sensitivity of the detection was more than 1 μM. The level of bilirubin in the GF was measured using the Bilirubin Assay Kit, which has a detection sensitivity of >0.5 mg/dL (Cell Biolabs, San Diego, California, USA).
PCR amplification of the 16S rRNA gene and next generation sequencing
Bacterial DNA was extracted from the GF using an Ultra Clean Soil DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, California, USA) in accordance with the manufacturer's instruction. The hypervariable V3-V4 region of 16S rDNA was amplified by PCR with 341f7 and R8068 primers. PCR was performed in accordance with the method reported by Takahashi et al.9 Sequencing was conducted using a paired-end and modified to 2×300 bp cycle run on an Illumina MiSeq sequencing system (Illumina, San Diego, California, USA) and MiSeq Reagent Kit V.3 chemistry.
16S rDNA-based operational taxonomic unit (OTU) analysis
The paired-end reads for each sample were joined using the Fastiq-Join method10 and processed with quality filtering.11 The average numbers (SD) of quality filter-passed reads were 37 202 (4848), 31 348 (4745) and 27 400 (2920) in the HC, FD before treatment and FD after treatment groups, respectively. The high-quality reads were then sorted and grouped into OTUs using the Quantitative Insights Into Microbial Ecology (QIIME) pipeline12 with default settings. Next, the reads were clustered into OTUs at 97% pairwise identity. To analyse the bacterial composition, TechnoSuruga Laboratory Microbial Identification Database DB-BA 10.0 (TechnoSuruga Laboratory, Shizuoka-City, Japan) was used in accordance with the method reported by Hisada et al.13 In this composition analysis, we focused on the genera representing >0.1% of the total GF microbiota, which was ∼ >85% in this study. Hierarchical clustering based on the unweighted pair group method with arithmetic mean (UPGMA) was performed using JMP V.8.0 (SAS Institute, Cary, North Carolina, USA) to analyse the overall bacterial community structure and the frequency of identification of each genus among the samples. The similarity between the bacterial communities was determined using Euclidean distance.
Statistical analysis
The Student's t-test or Mann-Whitney's U test was used to compare the bacterial abundance between HC and FD groups. The difference in the abundance in the FD group between before and after the treatment was examined by the Wilcoxon signed-rank test. The difference in the ratio of bacterial groups was examined by the χ2 for independence test. An analysis of similarity (ANOSIM) was used to test for statistical differences in bacterial community similarities using the vegan package (V.2.3-4) implemental in R (V.3.2.4). All p values were used two-sided and considered to be significant at p<0.05.
Results
Demographic and laboratory data
The FSSG scores for epigastric pain-like and postprandial distress-like symptoms were far greater in the FD before LG21 treatment than in the HC groups (table 1). The LG21 yogurt treatment significantly ameliorated both of these symptoms in the patients with FD. Among the GF-related parameters, the ratio of GF samples in which the bile acids were detectable was significantly greater in the FD before treatment than in the HC group. The median bile acids concentration was also significantly higher in the FD before treatment than in the HC groups. The ratio of bilirubin-positive samples also tended to be greater in the FD than in the HC group (p=0.061). The LG21 yogurt treatment exerted no significant effects on the level of bile acids or bilirubin in patients with FD.
Difference in the microbiota composition between FD before the treatment and HC groups
The rarefaction curves, which plots the OTU number as a function of the read number, showed that the contours of the FD before treatment and the HC groups almost overlapped (see online supplementary figure S1), suggesting no difference in the degree of bacterial species richness between them. The evaluation by Chao 1 index also drew the same conclusion.
supplementary data
OTUs generated from the 16S rDNA reads were then classified into phyla according to a similarity search (figure 1). The most dominant phylum was Firmicutes in both the FD before treatment and HC groups. However, the second/third major phyla were Bacteroidetes/Proteobacteria in the FD group and Proteobacteria/Bacteroidetes in the HC group, respectively. Indeed, the average of the relative abundance of Bacteroidetes/Proteobacteria was 24.2/4.9% in the FD group, while it was 13.5/29.5% in the HC group (table 2). Also in the comparison of the Bacteroidetes:Proteobacteria ratio, the number of subjects who had the Bacteroidetes > Proteobacteria abundance was 19 out of 24 FD subjects, whereas that of subjects who had such abundance was just 2 out of 21 HC subjects, in which the difference in the ratio between two groups was statistically significant. When the Bacteroidetes > Proteobacteria pattern was used as the criterion for diagnosing FD, the sensitivity and specificity were 79.2% and 90.4%, respectively. In the analysis at the genus and species levels, the abundance of genus Bacteroides and species B. dorei, belonging to the phylum Bacteroidetes, was also far greater in the FD than in the HC groups. Another interesting finding in the composition analysis was that the abundances of the phylum Acidobacteria and genus Edaphobacter that belonged to this phylum were far lower in the FD than in the HC groups. Furthermore, on comparing the ratio of subjects who had Acidobacteria (abundance>1%), the FD group had only 1 out of 24 subject, while the HC group had 16 out of 21 subjects, in which the difference was markedly significant. The sensitivity and specificity of the absence of Acidobacteria as a diagnostic criterion were 95.8% and 76.2%, respectively.
Predominant groups of bacteria
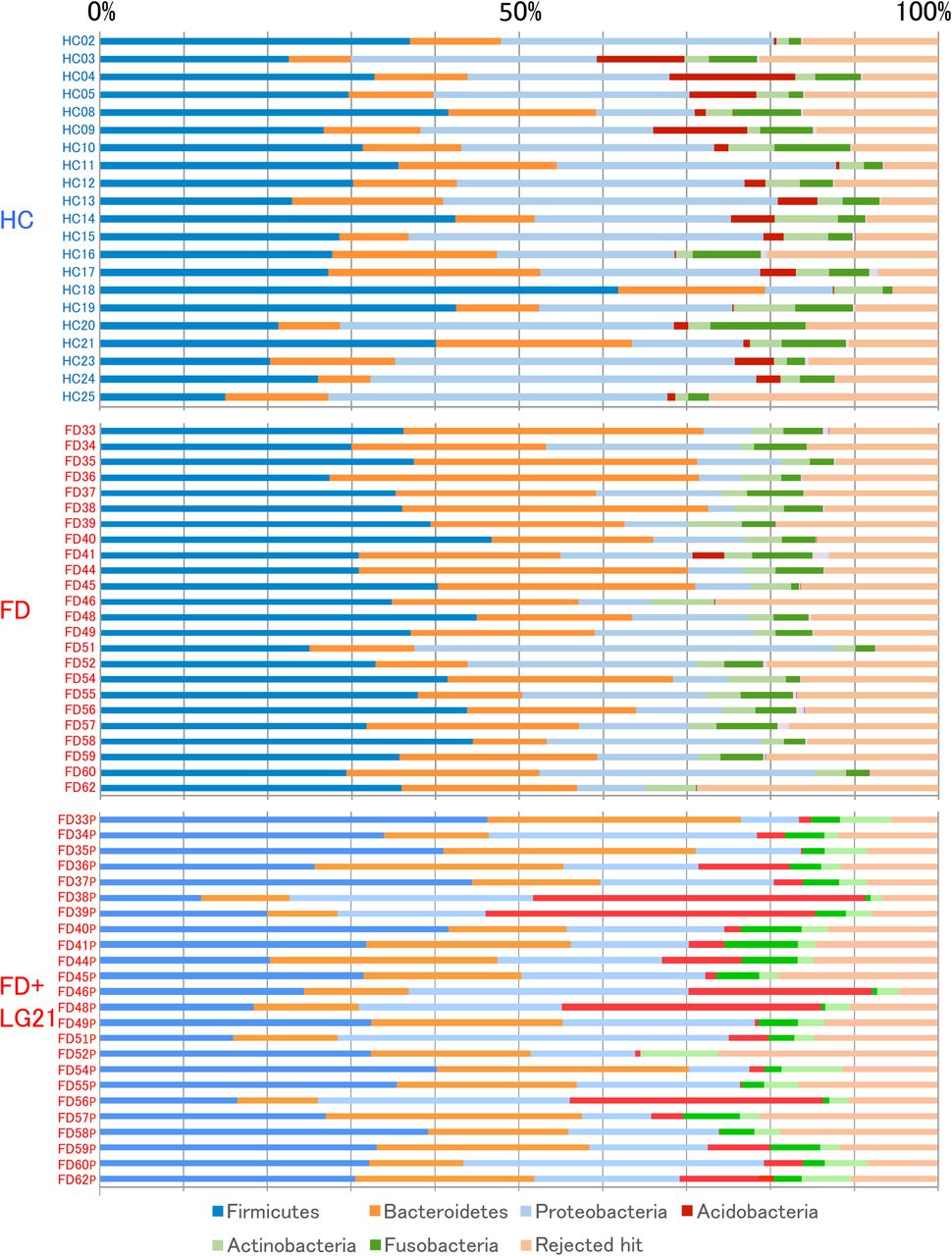
Bacterial composition at the phylum level. The relative abundance of the bacterial composition at the phylum level in each sample from the subjects in the HC, FD and FD after LG21 yogurt treatment groups is shown on a bar chart. The ID of each subject is tagged to the left of the bar. FD, functional dyspepsia; HC, healthy control.
Of further note, in the FD group, the bacterial species that predominantly inhabit the intestine, such as Escherichia/Shigella and Bifidobacterium longum were significantly higher than those in the HC group (table 2). Bile acids are known to selectively accelerate the growth of some Gram-negative bacteria like Escherichia/Shigella.14 Whereas the bile acids levels were significantly higher in the FD than in the HC groups, no significant correlation was observed between the concentration of bile acids and the abundance of those bacteria in each sample by Spearman's correlation coefficient by rank test (rs=0.031, p=0.93).
Interindividual bacterial variation at the genus level
The difference in the bacterial composition between the FD before treatment and the HC groups was also demonstrated by a hierarchical clustering analysis (figure 2, right side). Indeed, the Euclidian distance was significantly different between these two groups when examined by the ANOSIM test (R=0.41, p<0.01), indicating a definite difference in the overall bacterial community structure between them. In addition, the phylogenetic tree was roughly composed of a dominant cluster that largely consisted of HC subjects (indicated by a frame on the left side) and the other clusters that exclusively consisted of FD subjects. Furthermore, it appeared that the structure of the microbiota was different even in the FD subjects between those involved in the dominant cluster and those involved in the other clusters. Indeed, FDs 58, 51, 55 and 52 in the dominant cluster (indicated by arrows) expressed a Proteobacteria > Bacteroidetes phylum pattern that was typical of HC subjects, despite being enrolled as patients with FD.

Double-hierarchical clustering analysis. The samples from 45 subjects and 83 dominant genera are represented on a double-hierarchical clustering heat map. The blue and red squares represent lower and higher abundances, respectively. The clusters on the right side indicate the similarity among the individual (IDs on the left side) profiles at the genus level. A frame at the left represent the samples forming a dominant cluster. Open arrows indicate the FD subjects with samples showing the Bacteroidetes < Proteobacteria phylum pattern. The clusters at the bottom indicate the genera showing similarity in the frequency of identification among samples. See supplementary figures S2 and S3 to view the names of the genera in this figure.
Next, we examined the similarity in the frequency of identification among samples in the clustering of genera (figure 2, bottom side) to analyse the mechanism for the difference in the composition of microbiota between the FD and the HC groups. As a result, we found two distinct clusters of genera. One cluster of them was formed by a particular group that consisted of 32 genera (see online supplementary figure S2), including typical inhabitants in the intestine such as Bifidobacterium, Faecalibacterium and Bacteroides. The other cluster was formed by 51 genera (see online supplementary figure S3), including typical inhabitants in the stomach such as Neisseria, Haemophilus and Streptococcus. In eight FD subjects (FD 33, 36, 39, 45, 46, 54, 56 and 62), those 32 genera were particularly dominant, appeared as deep-red on a heat map, but such typical stomach-type genera among the 51 genera were scarce. Furthermore, all of eight subjects also showed a characteristic FD-type phylum profile (Bacteroidetes > Proteobacteria, absence of Acidobacteria), which were thus categorised as an FD subtype bearing lots of intestinal bacteria in the stomach.
Effect of LG21 yogurt treatment on the GF microbiota in patients with FD
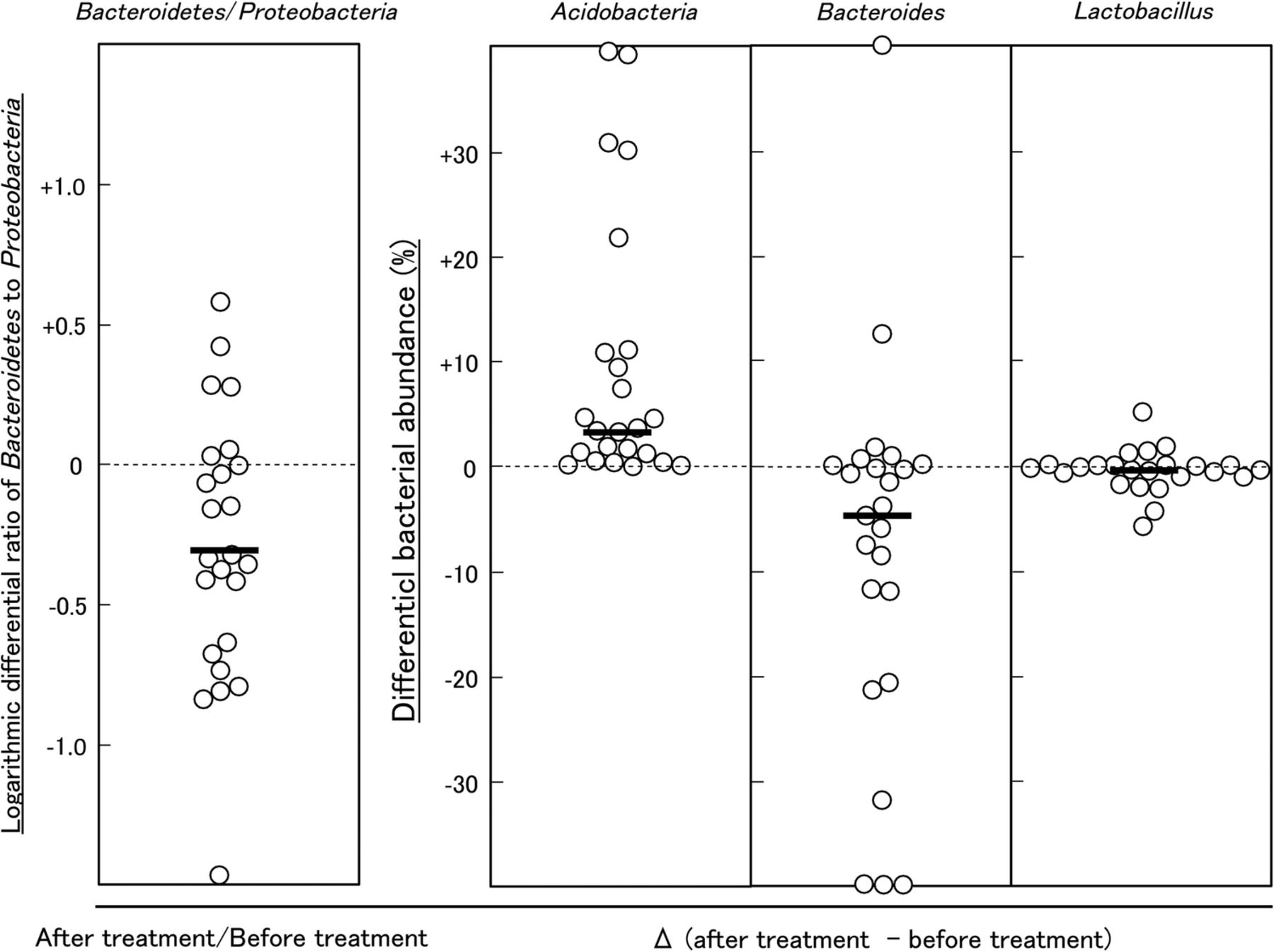
The treatment of patients with FD with LG21 yogurt restored the microbiota profile to that found in HC volunteers (figure 1 and table 2). First, the dominance of Bacteroidetes over Proteobacteria was markedly weakened after the treatment. Indeed, the number of subjects who had the Bacteroidetes > Proteobacteria abundance decreased to 12 patients after the treatment, compared with 19 before the treatment. Furthermore, the number of subjects whose Bacteroidetes:Proteobacteria ratio decreased after the treatment was as many as 18 out of 24 patients (figure 3). Second, both the prevalence and abundance of Acidobacteria markedly increased after the treatment. The number of subjects who had Acidobacteria (abundance>1%) markedly increased to 19 after the treatment, compared with just one before the treatment. A marked decrease in the abundance of Bacteroides was also a noteworthy effect of the treatment (figure 3), as the predominance of this genus was one of the major features of the change of bacterial composition found in patients with FD. Third, interindividual variation analysis demonstrated a disappearance of intestinal-type genera after LG21 treatment in seven out of the eight subjects whose GF microbiota had been densely inhabited with these genera (see online supplementary figure S4). Of note, although 109 CFUs of LG21 strain, which belongs to Lactobacillus, were administered to patients with FD every day for 12 weeks, the abundance of this genus in the GF did not increase after the treatment (figure 3).

{kind=link}
{kind=link}
{kind=link}
Effect of LG21 treatment on the bacterial ratio and abundance in patients with FD. Each symbol represents the differential ratio of Bacteroidetes:Proteobacteria, or abundance of Acidobacteria, Bacteroides and Lactobacillus in the patients with FD before and after LG21 treatment. Bars represent the median. FD, functional dyspepsia.
Discussion
In the present study, a greater increase in the ratio of bile acid-positive GF samples was found in the patients with FD than in the HC volunteers, although these FD subjects had not suffered from any organic or metabolic diseases that affected bile acid homeostasis. The reflux of fluid including bile acids from the duodenum into the stomach physiologically occurs during gastric motility.15 In healthy people, however, the stomach contained minimal amount of bile acids but lots of bicarbonates, because the phase III migrating motor complex plays a role in evacuating the bile acids from the stomach.16 Therefore, patients with FD whose GF were contaminated with a considerable amount of bile acids may have suffered from disturbance in the gastric motility.
A comprehensive analysis of the GF microbiota in the present study through bacterial 16S rDNA profiling demonstrated obvious change in the GF microbiota of patients with FD compared with that of HC volunteers. The bacterial composition was revealed to be clearly different even at the phylum level between GF and HC groups; the GF microbiota of the FD group was characterised by a Bacteroidetes > Proteobacteria abundance and the absence of Acidobacteria (FD type), while that of the HC group was characterised by a Bacteroidetes < Proteobacteria abundance and the presence of Acidobacteria (HC type). Furthermore, those changes in the GF microbiota in patients with FD were significantly resolved after LG21 yogurt treatment. Given that the changes in the GF microbiota paralleled the absence (HC), presence (FD) and improvement (FD after treatment) of symptoms, the change in the gastric microbiota was suggested to be involved in the pathophysiology underlying FD.
The dominance of Bacteroidetes over Proteobacteria in the FD group might be due to back transfer of the resident bacteria in the proximal small intestine accompanied by the reflux of the duodenal fluid, as the abundance of genus Bacteroides, a major member of the phylum Bacteroidetes and exclusively inhabits the intestine, was far greater in the FD than in the HC groups. A significantly greater abundance of typical intestinal inhabitants, such as Escherichia/Shigella and Bifidobacterium longum in the FD group, supports this idea. Disturbance in the gastric motility, such as emptying failure, might result in a long dwelling time for intestinal bacteria in the stomach.
Genus Edaphobacter, a member of phylum Acidobacteria, appeared to represent almost all of the bacteria of this phylum in the present study, because their levels were roughly the same. Thus far, for this genus, the isolation of two species (E. modestus and E. aggregans from alpine and forest soils, respectively) has been reported.17 Their optimal conditions for growth were 30°C and pH 5.5. While the biological significance of the colonisation by this genus in the stomach remains unclear, the acidophilic growth condition of the bacteria may have allowed them to colonise the stomach. However, it appeared unlikely that disappearance of Acidobacteria in the GF of patients with FD was due to a change in the acidity of GF, as the pH value in the FD before the treatment was not different from that in the HC. A change in the bacterial ecology in the stomach of patients with FD might result in the disappearance of Acidobacteria, as the recovery of this phylum occurred in parallel with a decrease in the abundance of Bacteroidetes in the patients after LG21 treatment.
In the analysis of individual bacterial profiles at the genus level, the GF microbiota from eight FD subjects had a unique bacterial community characterised by the dominance of intestinal-type bacteria included in the 32 genera but few of the stomach-type bacteria included in the 51 genera. Such marked accumulation of intestinal-type genera in these eight subjects strongly suggested back transfer of those bacteria in a group from the intestine to the stomach and not the growth of individual bacteria as the 32 genera formed a distinct cluster with dense linkage in the analysis for the frequency of identification. Small intestinal bacterial overgrowth (SIBO) is broadly defined as an increase in the number of bacteria in the proximal small intestine.18 More than 60% of Japanese patients with FD have been reported to have overlapping IBS, in which SIBO was considered a critical aetiological factor.19 Thus, a considerable number of patients with FD, especially those eight patients, in the present study might have had SIBO, which might have caused the migration of large numbers of intestinal-type bacteria to the stomach through the reflux of duodenal fluids.
Validated end points that may serve as objective biomarkers for the diagnosis and treatment of FD are needed. Our finding of an FD-type phylum profile may improve the accuracy of the diagnosis of FD and categorising FD subgroups. Indeed, the use of the FD-type phylum profile to distinguish the FD from the HC was revealed to have such high sensitivity and specificity in the present study that they were found to be especially effective for identifying a subtype of FD that is associated with the bacterial composition change in the stomach. Clear restoration of the FD-type profile to the HC-type profile in the FD groups after successful treatment with LG21 also proved this bacterial phenomenon to be an effective biomarker for the evaluation of the therapeutic effect.
In our previous study using a quantitative PCR and 16S rRNA gene sequencing, the bacterial count in the GF was as high as 108–109/mL.20 While many of them were supposed to be recently dead, inactivated or unculturable, the number was far higher than the count obtained by traditional culturing methods. Furthermore, the species richness (αdiversity) of the GF microbiota was as high as that of the faecal microbiota. These results suggested that the mass size and diversity of GF microbiota was great enough to significantly affect the pathophysiology of the stomach through the metabolites and components of the bacteria. While the linkage of the alteration in the GF microbiota in the pathophysiology underlying FD remains to be clarified, the gastric mucosa might suffer from disturbance when exposed to toxic intestinal bacterial cell components like lipopolysaccharides that stimulated leukocytes to generate pro-inflammatory cytokines.21 Such activation in the mucosal innate immune responses increases the mucosal permeability, which may lead to the malfunction of the gastric nervous system regulating gastric motility. Bile acids are also known to exert toxic effects on gastric as well as duodenal mucosa.22 Therefore, both of these kinds of toxic substances in the GF were suggested to be linked to the pathogenesis and pathophysiology of FD. Low-grade duodenal inflammation has been observed and proposed as an important pathophysiological mechanism in patients with FD.2 Recently, Vanheel et al reported that the patients with FD displayed increased duodenal mucosal permeability that potentially resulted in mucosal inflammation.23 Given that lipopolysaccharides and bile acids induce an acceleration in the mucosal permeability, the inflammation in the duodenal region in the patients with FD might be caused by the refluxed fluid including such potentially toxic substances. It is thought that probiotics is effective in the treatment of FD through reduction in the abundance of Escherichia/Shigella, a major source of toxic lipopolysaccharides, in the upper GI tract as well as restoration of the change in the gastric microbiota. The limitations associated with the present study include the relative small number of samples and absence of the samples from the HC volunteers after LG21 yogurt treatment.
References
Footnotes
Contributors MI and HN recruited the patients with FD and healthy control volunteers, had interview with them, and collected the clinical samples. TM, ST, TH and JT performed biochemical, bacteriological, genetic and statistical analyses. YK was involved in the study concept and design, data interpretation and drafting the manuscript.
Funding Meiji Co., Ltd. (Tokyo, Japan) financially supported this study.
Disclaimer The funder was not involved in the study design, data interpretation or drafting the manuscript.
Competing interests None declared.
Ethics approval The Ethics Committee Tokai University Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.